用於富氫氣氛中優先氧化CO的寬溫催化劑的製備方法及其產品和應用與流程
2023-06-01 17:08:21 2
本發明涉及用於富氫氣氛中優先氧化co的金屬催化劑及其製備方法和應用。
背景技術:
近年來,清潔能源愈來愈受到人們的重視。其中,燃料電池是一種環境友好型的發電裝置。質子交換膜燃料電池(pemfc)是一種非常理想的燃料電池,它具有體積小、重量輕、零排放、能量轉換密度高等特點,在未來靜態裝置、氫燃料電池汽車、軍事設備、航空航天等領域具有廣闊的應用前景。燃料電池的氫源主要來源於甲醇和天然氣等碳氫化合物的水蒸汽重整、水煤氣變換反應等,該類氫源通常含有大約0.5%~2%的微量co。co極易吸附在pemfc的pt電極表面,使電極毒化,嚴重降低電池性能。因此,氫源在進入燃料電池前,必須首先進行co淨化處理,把co的含量控制在10ppm以下。為解決該問題,科研人員嘗試了很多方法。目前去除燃料電池的所用氫源中co的方法主要有吸附法、pd膜分離法、co甲烷化法和co選擇性氧化法。經對比,人們發現co選擇性氧化是去除該類氫源中微量co的最理想方法。
目前,用於富氫氣氛co優先氧化(下文中以prox表示)的催化劑主要分為以下幾種:
(a)金基催化劑:負載型au催化劑由於具有極高的低溫co氧化活性,因而受到了人們極大的關注。然而由於h2氧化反應的競爭,人們發現隨著反應溫度升高,co氧化選擇性迅速降低,很難將co濃度降到10ppm以下。例如,中國專利申請cn102441401a中提到將au納米顆粒負載在銅-鈦混合氧化物載體上,au負載量為0.5~5wt%,co100%轉化的溫度區間僅為30-60℃,工作溫度局限性較大。另外,au催化劑的穩定性相對較差,而且其活性對製備方法與過程的要求非常嚴格,使得au催化劑 在prox反應中的應用受到了較大限制。
(b)銅基非貴金屬催化劑:該類催化劑和其它類型的催化劑相比,雖然成本較低,但是該類催化劑在prox反應中,通常反應溫度達到100℃以上才會有活性,而且在co2和水汽存在的條件下穩定性差。如中國專利申請cn102407123a中提及的cuo擔載的ceo2催化劑。
(c)鉑族貴金屬催化劑:目前單一pt族金屬(pt、ru、rh、pd、ir)催化劑在100℃以下活性很差(j.phys.chem.b2005,109,23430-23443),通常需要以雙金屬合金或添加助劑的方式來提高催化活性。如新日本石油株式會社提出以ptru雙金屬合金作為活性組分的催化劑(參見cn101507924a)。另外,中國專利申請cn101856621提出了以鉑族金屬為活性成分,並以等體積浸漬法添加fe、co、ni等過渡金屬作為助劑的催化劑,它們能夠在60~100℃的溫度範圍內將氫氣中的co完全脫除。中國專利申請cn101428227a提出了一種利用分步或共浸漬的方法製備的銥基雙組份催化劑,其中助劑為fe、sn、mn、co、ni、cr、和/或zn,該催化劑可以在60~100℃的溫度範圍內將氫氣中的co高選擇性轉化。日本的watanabe研究小組利用離子交換法,在mordenite沸石上沉積了不同pt:fe質量比例的pt-fe/zeolite合金催化劑。他們製備的催化劑能夠在85-150℃的溫度範圍內實現將氫氣中的co高選擇性轉化(appl.catal.b:environ.2003,46,595–600)。中科院大連化學物理研究所的包信和課題組利用連續浸漬法和共浸漬法,分別獲得了雙組份pt-fe/sio2,pt-fe/炭黑催化劑樣品,然而這些催化劑僅僅可以在25~50℃的狹窄溫度範圍內將氫氣中的co高選擇性轉化(science,2005328,1141-1144,energyenviron.sci.,2012,5,6313–6320)。在他們的工作中,他們首次指出以氧化物為助劑的pt基催化劑,co氧化的活性中心為pt-氧化物助劑的界面(science,2005328,1141-1144,acc.chem.res.,2013,46,1692–1701);廈門大學的鄭南峰老師課題組也同樣指出,pt-fe催化劑體系的co氧化的活性中心為pt-fe(oh)3助劑的界面。然而,在這些研究中,氧化物助劑的添加是通過使用浸漬法、沉澱法、等液相方法實現的,這些方法無法實現催化劑中貴金屬活性組分-氧化物助劑界面的最佳優化,抑制了催化劑性能的有效提高,導致co完全轉化的工作溫度區間窄,無法滿足實際應用要求。
另外,自原子層沉積技術問世以來(us4,058,430(1977)),利用原子層沉積精確控制的技術優勢嘗試催化劑製備已獲得了人們的關注(surf.sci.rep.,(2016)doi:10.1016/j.surfrep.2016.03.003;acc.chem.res.,2013,46,1806–1815;acscatal.2015,5,1804-1825)。其中,利用原子層沉積技術對金屬催化劑進行氧化物包裹,進而實現催化劑性能的調控,也已有一些相關文獻報導例如:利用原子層沉積技術在pd/al2o3催化劑表面沉積al2o3包裹層,實現催化劑在高溫乙烷部分氧化脫氫反應中的抗燒結和抗積炭性能(science,2012,335,1205-1208;pct/us2012/039343);利用子層沉積技術在cu/γ-al2o3催化劑表面沉積al2o3包裹層,防止cu活性組分在液相催化反應中的浸出(angew.chem.2013,125,14053–14057);利用原子層沉積技術在co/c催化劑表面沉積tio2包裹層,實現催化劑在電催化反映中活性的提高(acscatal.2015,5,3463-3469);利用原子層沉積技術在pd/al2o3催化劑表面沉積zro2包裹層,實現催化劑在甲烷完全燃燒反應中的穩定性和催化活性的提高(acscatal.2015,5,5696-5701)等。上述有關利用原子層沉積技術對金屬催化劑進行氧化物包裹的工作,主要側重通過氧化物包裹層的物理阻隔層,以解決金屬顆粒在催化反應過程中的燒結問題,沉積的氧化包裹層較厚(大於1nm)以達到穩定性提高的效果,氧化物包裹的金屬顆粒是通過其氧化物層內的微孔而參與催化反應的。然而,在金屬顆粒上沉積高分散性的氧化物物種,甚至氧化物單體(m1ox,m是金屬原子)物種,從而實現金屬-氧化物界面的最大優化還無相關研究。此外,到目前為止,利用化學氣相沉積或原子層沉積技術製備催化劑,更是沒有在富氫氣氛中優先氧化co反應中應用的相關報導。
技術實現要素:
為了克服現有技術中的一個或多個缺陷,本發明的目的是提供一種在富氫氣氛下優先氧化co的寬溫催化劑,其可以在寬溫度範圍(例如可在-80~200℃的溫度範圍)內使用,並且能夠在富氫氣氛co優先氧化反應中表現出優異的催化活性、選擇性和穩定性。
為此,在一方面,本發明提供一種用於在富氫氣氛中優先氧化co的寬溫催化劑的方法,其特徵在於,所述催化劑包括載體、活性組分和助劑, 其中所述載體是選自sio2、al2o3、tio2、mgo、ceo2、zro2、活性炭、炭黑、石墨烯和碳納米管中的一種或多種;所述活性組分是選自pt、ir、ru、rh和pd中的一種或多種,其在所述寬溫催化劑中的含量為0.1~10wt%;所述助劑是選自鐵氧化物、鈷氧化物和鎳氧化物中的一種或多種,其以金屬元素計在所述寬溫催化劑中的含量為0.01~15wt%,
所述方法包括:
提供包括所述活性組分和所述載體的負載型催化劑前驅體;
通過化學氣相沉積法或原子層沉積法將所述助劑沉積到所述負載型催化劑前驅體的表面上,從而得到所述寬溫催化劑。
優選地,所述助劑的沉積包括以下步驟:
(a)將所述負載型催化劑前驅體放置在處於20~500℃的反應器中,並引入作為助劑前驅體的鐵前驅體、鈷前驅體和/或鎳前驅體的蒸汽以吸附到所述負載型催化劑前驅體的表面上;
(b)引入氧化劑或還原劑以使吸附在所述負載型催化劑前驅體的表面上的所述助劑前驅體轉化為所述助劑;
(c)任選地相繼或同時重複執行以上步驟(a)和(b)一次或多次以調控所述助劑的質量含量。
優選地,所述鐵前驅體是選自二茂鐵、乙烯基二茂鐵、乙基二茂鐵、氨基二茂鐵、二甲氨基二茂鐵、乙醯丙酮鐵、雙(2,4-二甲基戊二烯基)鐵、、(2,2,6,6-四甲基-3,5-庚二酮酸)鐵(iii)、雙(n,n′-二叔丁基乙脒基)鐵、羰基鐵和叔丁醇鐵中的一種或多種;最優選地,所述鐵前驅體是選自二茂鐵;
所述鈷前驅體是選自二茂鈷、乙醯丙酮鈷、雙(n,n′-二異丙基乙脒基)鈷、二羰基環戊二烯鈷、叔丁基三羰基鈷、(2,2,6,6-四甲基-3,5-庚二酮酸)鈷和2-甲氧基乙醇鈷中的一種或多種;最優選地,所述鈷前驅體是選自二茂鈷;
所述鎳前驅體是選自二茂鎳、乙醯丙酮鎳、雙(n,n′-二異丙基乙脒基)鎳、(2,2,6,6-四甲基-3,5-庚二酮酸)鎳(ii)、二丁基二硫代氨基甲酸鎳(ii)和2-甲氧基乙醇鎳中的一種或多種。
優選地,所述氧化劑是選自o2、o3、h2o、h2o2、no和no2中的一種或多種;更優選地,所述氧化劑是選自o2、o3、h2o、h2o2;所述 還原劑是選自h2、nh3和n2h4中的一種或多種;更有選地,所述還原劑是選自h2。
優選地,所述方法還包括以下步驟:在所述步驟(a)和步驟(b)之間以及在所述步驟(b)之後,利用惰性氣體吹掃所述反應器。
優選地,所述步驟(a)和(b)相繼重複執行1-10次。
優選地,所述負載型催化劑前驅體通過商購獲得,或者通過浸漬法將所需量的活性組分的可溶性鹽負載到載體上然後經過乾燥焙燒獲得。
在另一方面,本發明提供一種用於在富氫氣氛中優先氧化co的寬溫催化劑,其特徵在於,所述催化劑包括載體、活性組分和助劑,
其中所述載體是選自sio2、al2o3、tio2、mgo、ceo2、zro2、活性炭、炭黑、石墨烯和碳納米管中的一種或多種;
所述活性組分是選自pt、ir、ru、rh和pd中的一種或多種,其在所述寬溫催化劑中的含量為0.1~10wt%;
所述助劑是選自鐵氧化物、鈷氧化物和鎳氧化物中的一種或多種,其以金屬元素計在所述寬溫催化劑中的含量為0.01~15wt%,
並且其中所述寬溫催化劑是通過化學氣相沉積法或原子層沉積法將助劑沉積到包括所述活性組分和所述載體的負載型催化劑前驅體的表面上獲得的,並且所述寬溫催化劑能夠實現在富氫氣氛中優先氧化co。
優選地,所述活性組分是選自pt;優選地,所述助劑是選自鐵氧化物;優選地,所述載體是選自sio2,並且所述寬溫催化劑能夠在-80~200℃的寬溫度範圍實現在富氫氣氛中優先氧化co。
在另一方面,本發明提供通過上述方法製備的寬溫催化劑或上述寬溫催化劑用於在富氫氣氛中優先氧化co的應用。
優選地,所述寬溫催化劑在使用之前進行預處理,其中所述預處理是在100~600℃溫度下首先用氧氣氧化0.5~5小時,然後用氫氣還原0.5~5小時。
優選地,所述寬溫催化劑在使用之前進行預處理,其中所述預處理是在150~300℃溫度下首先用氧氣氧化0.5~2小時,然後用氫氣還原0.5~2小時。
優選地,所述富氫氣氛中co和o2的體積比為1:0.5至1:2。
優選地,所述富氫氣氛中co和o2的體積比為1:0.5至1:1。
本發明通過化學氣相沉積法或原子層沉積法將金屬氧化物助劑沉積到pt族貴金屬(pt、ir、ru、rh或pd)負載型催化劑前驅體表面上而實現對催化劑的貴金屬活性組分-氧化物助劑界面的最佳優化。通過這樣的方法獲得的貴金屬催化劑在富氫氣氛中co優先氧化反應中能夠表現出優異的催化性能,並且能夠在比現有催化劑顯著更寬的溫度區間(例如可在-80~200℃的寬溫度區間)實現富氫氣氛中co的高選擇性、高轉化率氧化。此外,在水蒸氣和co2存在的情況下,所述催化劑也能夠長時間保持穩定。
附圖說明
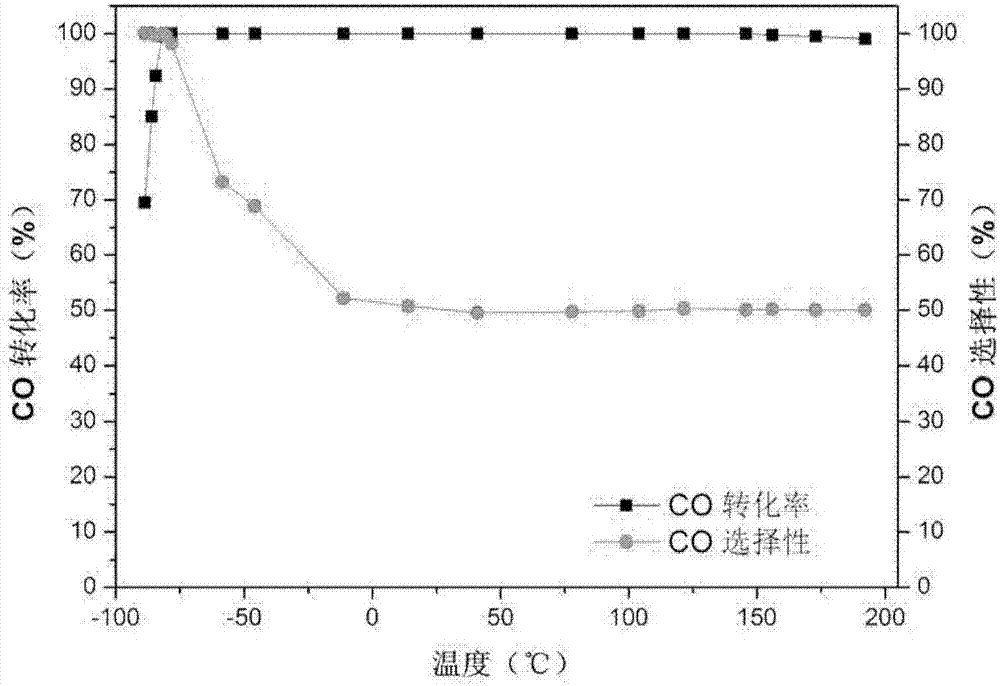
圖1示出了根據本發明實施例1製備的pt-1cfeox/sio2寬溫催化劑在富氫氣氛中優先氧化co的co轉化率和co選擇性的曲線圖,其中使用的富氫反應氣樣品中的co:o2:h2的體積比為1:0.5:48。
圖2示出了根據本發明實施例1製備的pt-1cfeox/sio2寬溫催化劑在富氫氣氛中優先氧化co的co轉化率和co選擇性的曲線圖,其中使用的富氫反應氣樣品中的co:o2:h2的體積比為1:1:48。
圖3示出了根據本發明實施例1製備的pt-1cfeox/sio2寬溫催化劑在80℃溫度下在反應氣樣品含有水蒸汽(h2o)和co2的情況下的穩定性曲線圖。
圖4示出了根據本發明實施例2製備的pt-5cfeox/sio2寬溫催化劑在富氫氣氛中優先氧化co的co轉化率和co選擇性的曲線圖,其中使用的反應氣樣品中的co:o2:h2的體積比為1:0.5:48。
圖5示出了根據本發明實施例9製備的pt-1cfeox/c寬溫催化劑在富氫氣氛中優先氧化co的co轉化率和co選擇性的曲線圖,其中使用的反應氣樣品中的co:o2:h2的體積比為1:1:48。
圖6示出了根據本發明實施例10製備的pt-1cfeox/tio2寬溫催化劑在富氫氣氛中優先氧化co的co轉化率和co選擇性的曲線圖,其中使用的反應氣樣品中的co:o2:h2的體積比為1:1:48。
圖7示出了根據本發明實施例11製備的pt-1cfeox/al2o3寬溫催化劑 在富氫氣氛中優先氧化co的co轉化率和co選擇性的曲線圖,其中使用的反應氣樣品中的co:o2:h2的體積比為1:1:48。
圖8示出了根據本發明實施例12製備的pt-1ccoox/sio2寬溫催化劑在富氫氣氛中優先氧化co的co轉化率和co選擇性的曲線圖,其中使用的反應氣樣品中的co:o2:h2的體積比為1:0.5:48。
圖9示出了根據本發明實施例19製備的pt-1cniox/sio2寬溫催化劑在富氫氣氛中優先氧化co的co轉化率和co選擇性的曲線圖,其中使用的反應氣樣品中的co:o2:h2的體積比為1:0.5:48。
圖10示出了根據本發明實施例25製備的ir-2cfeox/sio2寬溫催化劑在富氫氣氛中優先氧化co的co轉化率和co選擇性的曲線圖,其中使用的反應氣樣品中的co:o2:h2的體積比為1:1:48。
圖11示出了根據本發明實施例25製備的ir-2cfeox/sio2寬溫催化劑在80℃溫度下的穩定性曲線圖。
圖12示出了根據本發明實施例26製備的ir-1ccoox/sio2寬溫催化劑在富氫氣氛中優先氧化co的co轉化率和co選擇性的曲線圖,其中使用的反應氣樣品中的co:o2:h2的體積比為1:1:48。
圖13示出了根據本發明實施例27製備的pt-1cfeox/sio2(fe(acac)3)寬溫催化劑在富氫氣氛co優先氧化的co轉化率和co選擇性的曲線圖,其中使用的反應氣樣品中的co:o2:h2的體積比為1:0.5:48。
具體實施方式
為實現上述目的,本發明的發明人經過深入研究,出乎意料地發現,利用化學氣相沉積法或原子層沉積法,在pt族貴金屬(pt、ir、ru、rh或pd)負載型催化劑前驅體表面上可以實現精準沉積具有高分散性的氧化物助劑;而且通過利用化學氣相沉積,尤其是原子層沉積技術的精確控制優勢,能夠實現對催化劑的貴金屬活性組分-氧化物助劑界面的最佳優化,進而最大程度提高催化劑的活性和貴金屬的利用率,獲得了能夠在比現有催化劑顯著更寬的溫度區間(例如可在-80~200℃的寬溫度區間)實現富氫氣氛中co的高選擇性、高轉化率氧化的寬溫催化劑。
更具體地,本發明提供的用於富氫氣氛中優先氧化co的寬溫高性能 催化劑包括載體、貴金屬活性組分和非貴金屬氧化物助劑,其中的載體為氧化物或碳基材料;貴金屬活性組分為選自pt、ir、ru、rh和pd(即pt族貴金屬)的一種或多種;非貴金屬氧化物助劑為選自鐵氧化物(feox)、鈷氧化物(coox)和鎳氧化物(niox)(其中的下標x僅表示所形成的相應氧化物中o的原子係數,其對於本領域技術人員是熟知的,沒有特定值限制)中的一種或多種,並且其中,基於所述寬溫催化劑的總重量,所述貴金屬活性組分的含量為0.1~10wt%,優選1~5wt%,所述非貴金屬氧化物助劑以金屬元素計的含量為0.01~10wt%,優選0.1~10wt%。
在本發明中,對使用的pt族貴金屬負載型催化劑前驅體沒有特別限制,其可以是商購或商品負載型催化劑,也可以是通過本領域熟知的浸漬法、離子交換法、沉澱法、溶膠凝膠法等方法製備獲得的負載型催化劑前驅體。
在本發明中,對所使用的載體沒有特別要求,只要能夠負載上述貴金屬活性組分即可。常見的或優選的,使用的載體是選自sio2、al2o3、tio2、mgo、ceo2、zro2、活性炭、炭黑、石墨烯和碳納米管中的一種或多種,其中更優選sio2、al2o3、tio2、ceo2、zro2或活性炭,更優選sio2。
在本發明中,非貴金屬氧化物助劑是通過化學氣相沉積法或原子層沉積方法沉積到pt族貴金屬負載型催化劑前驅體的表面上的,不受限於任何理論。其中,一種成核生長理論是:由於pt族金屬顆粒通常具有高催化活性,當氧化物助劑的金屬前驅體被引入到pt族貴金屬負載型催化劑前驅體的表面上時,氧化物助劑的金屬前驅體通常通過解離吸附的方式吸附到pt族金屬顆粒表面;此後,利用氧化劑或還原劑可以把氧化物助劑的金屬前驅體配體有效去除,並形成pt族貴金屬活性組分--氧化物助劑界面。
優選地,所述非貴金屬氧化物助劑的沉積包括以下步驟:
(a)將所述負載型催化劑前驅體放入反應器或反應腔內,並加熱到適當的溫度(例如為20~500℃),然後引入適當劑量的助劑前驅體的蒸汽以吸附在所述負載型催化劑前驅體的表面上;
(b)引入適當劑量的氧化劑或還原劑(取決於所述助劑前驅體至所述氧化物助劑的轉化反應所需,即如果該轉化是氧化反應,則引入氧化劑;如果該轉化是還原反應,則引入還原劑),使氧化劑或還原劑與吸附在催 化劑前驅體表面上的物助劑前驅體發生化學反應,從而實現氧化物助劑在催化劑表面上的沉積,由此獲得所需寬溫催化劑
(c)任選地相繼或同時重複執行上述步驟(a)和(b)一次或多次(即沉積一個周期或多個周期),以調控氧化物助劑在所述寬溫催化劑中的含量。
更優選地,通過原子層沉積法實現氧化物助劑的沉積或添加。更優選地,其中的沉積包括以下步驟:
(1)將所述負載型催化劑前驅體放入反應器或反應腔內,並加熱到一定的溫度(例如為20~500℃),然後面引入適當劑量的助劑前驅體的蒸汽以吸附在所述負載型催化劑前驅體的表面上;
(2)任選地利用惰性氣吹掃所述反應器或反應腔,以將其中剩餘的助劑前驅體和其他反應產物吹掃乾淨;
(3)引入適當劑量的氧化劑或還原劑,使氧化劑或還原劑與吸附在催化劑前驅體表面上的助劑前驅體發生化學反應,從而實現氧化物助劑在催化劑表面上的可控沉積。
(4)任選地利用惰性氣體吹掃所述反應器或反應腔,以將其中剩餘的氧化劑或還原劑以及其他反應產物吹掃乾淨,由此獲得所述寬溫催化劑。
(5)任選地相繼重複執行上述步驟(1)~(4)一次或多次(即沉積一個周期或多個周期),以調控氧化物助劑在在所述寬溫催化劑中的含量。
在本發明中,氧化物助劑的沉積期間,可以僅沉積一種氧化物助劑,也可以沉積兩種或三種氧化物助劑。而且,這些氧化物助劑可以分別在不同沉積周期中進行沉積,也可以同時在同一個或幾個沉積周期中進行沉積。
在本發明中,用於通過化學氣相法沉積或原子層沉積法沉積的助劑前驅體為如下:
作為鐵氧化物助劑的鐵前驅體,其優選是選自二茂鐵、乙烯基二茂鐵、乙基二茂鐵、氨基二茂鐵、二甲氨基二茂鐵、乙醯丙酮鐵、雙(2,4-二甲基戊二烯基)鐵、(2,2,6,6-四甲基-3,5-庚二酮酸)鐵(iii)、雙(n,n′-二叔丁基乙脒基)鐵、羰基鐵、和叔丁醇鐵中一種或多種。在這些之中,特別優選的鐵前驅體是二茂鐵。
作為鈷氧化物助劑的鈷前驅體,其優選是選自二茂鈷、乙醯丙酮鈷、雙(n,n′-二異丙基乙脒基)鈷、二羰基環戊二烯鈷、叔丁基三羰基鈷、(2,2,6,6-四甲基-3,5-庚二酮酸)鈷和2-甲氧基乙醇鈷中的一種或多種。在這些之中,特別優選的鈷前驅體是二茂鈷。
作為鎳氧化物助劑的鎳前驅體,其優先是選自二茂鎳、乙醯丙酮鎳、雙(n,n′-二異丙基乙脒基)鎳、(2,2,6,6-四甲基-3,5-庚二酮酸)鎳(ii)、二丁基二硫代氨基甲酸鎳(ii)和2-甲氧基乙醇鎳中的一種或多種。在這些之中,特別優選的鎳前驅體是二茂鎳。
在本發明中,氧化物助劑沉積中使用的氧化劑優選是選自o2、o3、h2o、h2o2、no和no2中的一種或多種,使用的還原劑優選是選自h2、nh3和n2h4中的一種或多種。其中更優選地,使用的氧化劑或還原劑選自o2、o3、h2o、h2o2、h2或nh3。
在本發明中,使用的惰性氣體是n2、ar、he或其組合,優選為n2。
在本發明中,在沉積氧化物助劑過程中,反應器或反應腔的加熱溫度或負載型催化劑前驅體的溫度優選為20~500℃,更優選為50~350℃,最優選為100~200℃。
在本發明中,對於沉積次數或沉積周期數沒有特別限制,可以根據需要進行一次或多次沉積。優選地,氧化物助劑的沉積次數1~50次,更優選為1~20次,最優選為1~10次。
在本發明中,對於所述寬溫催化劑應用的富氫氣氛的組成沒有特別限制。通常,在所述寬溫催化劑的應用中,富氫反應氣氛中co的含量小於5%,h2的含量大於10%,co和o2的體積比為在1:0.5~2的範圍內。優選地,針對用於氫燃料電池的氫源氣氛,其中co的含量小於5%,h2的含量大於30%,co和o2的體積比為在1:0.5~1的範圍內。
優選地,在使用本發明的寬溫催化劑之前,對該寬溫催化劑進行預處理。例如,預處理過程是在100~600℃溫度下用氫氣還原0.5~5小時。更優選地,預處理過程是在200~300℃溫度下用氫氣還原0.5~2小時。進一步優選地,預處理過程是在200~300℃溫度下用氫氣還原0.5~2小時,然後用富氫氣氛反應氣體在室溫~300℃溫度(例如100~200℃溫度)下再處理0.1~5小時(例如0.2~1小時)。
本發明的優點包括但不限於以下方面:
本發明提供的沉積氧化物助劑的方法的重複性好、適用性廣,適合於任何pt族貴金屬負載型催化劑,包括商購(商品)pt族貴金屬(pt、ir、ru、rh、pd)負載型催化劑,或者是通過浸漬法、離子交換法、沉澱法、溶膠凝膠法等製備的各種pt族貴金屬(pt、ir、ru、rh、pd)負載型催化劑。
本發明提供的沉積氧化物助劑的方法操作簡單,僅僅需要化學氣相沉積法或者原子層沉積法一步操作,便可實現pt族貴金屬(pt、ir、ru、rh、pd)負載型催化劑性能的極大提高。
本發明提供的氧化物助劑添加方法中,所用的氧化物助劑的金屬有機前驅體價格低廉,能夠以較低的成本實現催化劑性能的極大提高。
本發明利用化學氣相沉積法,尤其是原子層沉積法,在傳統的pt族貴金屬(pt、ir、ru、rh、pd)負載型催化劑表面上沉積氧化物助劑後,所獲得的催化劑可以在寬溫區間(例如可在-80~200℃溫度區間)實現富氫氣氛中co的高選擇性、高轉化率氧化,這樣的工作溫度範圍是迄今適用工作溫度範圍最寬的;此外,本發明的寬溫催化劑可以在水蒸氣和co2存在的情況下,能夠長時間保持穩定。
下面通過實施例進一步說明本發明,但本發明並不限於以下實施例。實施例1:pt-1cfeox/sio2催化劑的製備及其在富氫氣氛中優先氧化co的活性測試
pt/sio2催化劑前驅體的製備:利用原子層沉積法獲取的。使用粘滯流動型原子層沉積反應器(arradiance),原子層沉積的溫度為250℃,實驗的pt前驅體為(三甲基)甲基環戊二烯合鉑(iv)(mecpptme3,stremchemicals),利用加熱套,把該金屬前驅體源容器的溫度加熱到70℃,以獲得足夠的mecpptme3前驅體蒸汽壓。氧化劑為高純o2(99.999%,南京特種氣體),惰性氣體為高純n2(99.999%,南京特種氣體)。在催化劑製備過程中,把700mgsio2載體(300m2/g,alfaaesar)放入其反應腔內,(a)利用高純n2把mecpptme3的蒸氣引入到反應腔內,時間為10分鐘;(b)把mecpptme3源停止後,用高純n2氣吹掃5分鐘;(c)引高純o2 進入反應腔內,時間為3分鐘;(d)然後再關掉o2源,並用高純n2氣吹掃5分鐘。重複上述步驟(a-d)2次,即做2個原子層沉積周期。從反應腔內取出樣品,獲得pt/sio2催化劑前驅體。其中pt的質量含量為3.6wt%,根據高分辨電鏡結果,pt顆粒尺寸為2.7±0.4nm。
feox助劑的沉積:通過電阻加熱,把粘滯流動型原子層沉積反應設備(arradiance)的反應腔溫度加熱到120℃,使用的feox助劑的金屬前驅體是二茂鐵(fecp2,sigmaaldrich),利用加熱套,把該金屬前驅體源容器的溫度加熱到90℃,以獲得足夠的二茂鐵前驅體蒸汽壓。將500mgpt/sio2催化劑前驅體樣品放置到原子層沉積反應腔內。打開二茂鐵容器和原子層沉積反應腔之間的隔離閥門,二茂鐵蒸汽混入高純n2(99.999%,南京特種氣體),並被高純n2引入到原子層沉積反應腔內。二茂鐵並在pt/sio2催化劑前驅體中的pt顆粒表面發生解離吸附,繼而實現fe在pt表面的成核生成,引入時間為5分鐘。關閉二茂鐵源後,引入作為氧化劑的高純o2(99.999%,南京特種氣體)至反應腔內,時間為3.3分鐘,以將在pt表面解離吸附的二茂鐵的有機配體部分通過氧化燃燒掉,並轉化為feox,從而實現feox在催化劑表面的沉積。
最後,從反應腔內取出樣品,獲得pt-1cfeox/sio2寬溫催化劑(其pt的質量含量仍為3.6wt%),feox助劑以鐵元素計的質量含量是0.1wt%。
活性測試1:進行富氫氣氛中優先氧化co的活性測試。將實施例1中獲得的pt-1cfeox/sio2催化劑100mg首先與1g石英砂研磨均勻混合(以防止反應中形成「熱點」),反應器為u-型石英管(自製);催化劑預處理:首先在10%o2/he氣氛中在200℃處理1小時,隨後切換為10%h2/he,繼續處理2小時,最後用1%co+0.5%o2+48%h2+50.5%he的反應氣體再處理20分鐘。活性測試的反應氣體樣品的組成為1%co+0.5%o2+48%h2+50.5%he,反應氣的流速為60ml/min。並在-80~200℃溫度區間內對催化劑進行活性測試,測試結果如圖1所示,從該圖1中可以看出,利用本發明製備的寬溫催化劑,能夠在-80~200℃溫度區間內實現富氫氣氛中co的高轉化率轉化。
活性測試2:類似於活性測試1中的過程,將上述pt-1cfeox/sio2催化劑進行富氫氣氛中優先氧化co的活性測試,其中催化劑的用量和預處理過程同上所述,只是將反應氣體樣品的組成調整為1%co+1%o2+48%h2+50%he,反應氣的流速為60ml/min。活性測試結果如圖2所示,從該圖2中可以看出,利用本發明製備的寬溫催化劑,能夠在-80~200℃溫度區間內實現富氫氣氛中co的優選完全轉化。
穩定性測試:對上述pt-1cfeox/sio2催化劑進行穩定性測試,其中催化劑的用量和預處理過程如活性測試1中所述。穩定性測試中反應氣的總流量為60ml/min,氣體的組成為1%co+0.5%o2+48%h2+3%h2o+20%co2+27.5%he(模擬燃料電池中的富氫氣氛組成),把反應溫度保持在80℃維持不變,對樣品連續測試160小時。穩定性測試結果如圖3所示,從該圖3可以看出,所述催化劑對於含有水和co2的富氫氣氛,能夠長時間保持活性而無明顯失活現象。
實施例2:pt-5cfeox/sio2催化劑製備以及富氫氣氛中co優先氧化反應活性
pt/sio2催化劑前驅體的製備:首先以矽酸四乙酯(teos)為原料,通過鹼性水解的方法合成單分散二氧化矽小球(sio2)。其次將合成的二氧化矽小球進行300℃的高溫煅燒處理。此後,用3-氨基丙基-三乙氧基矽烷(atpes)對煅燒後的二氧化矽小球表面進行修飾(atpes-sio2)。將1.4g的atpes-sio2和7.9ml的氯鉑酸加入30ml乙醇中,室溫下攪拌24小時。離心烘乾,並在10%h2/ar氣氛中350℃條件下處理2小時,得pt/sio2(wi)。其中,催化劑中pt的質量含量是3.2wt%,pt顆粒尺寸約為3nm。
feox助劑的沉積:通過電阻加熱,把粘滯流動型原子層沉積反應設備(arradiance)的反應腔溫度加熱到120℃,使用的feox助劑的金屬前驅體是二茂鐵(fecp2,sigmaaldrich),利用加熱套,把該金屬前驅體源容器的溫度加熱到90℃,以獲得足夠的二茂鐵前驅體蒸汽壓。將150mgpt/sio2催化劑前驅體樣品放置到原子層沉積反應腔內。(a)打開二茂鐵容器和原子層沉積反應腔之間的隔離閥門,二茂鐵蒸汽混入高純n2(99.999%,南 京特種氣體),並被高純n2引入到原子層沉積反應腔內。二茂鐵並在pt/sio2催化劑前驅體中的pt顆粒表面發生解離吸附,繼而實現fe在pt表面的成核生成,引入時間為2.5分鐘。(b)關閉二茂鐵源後,並用高純n2氣繼續吹掃5分鐘,(c)引入作為氧化劑的高純o2(99.999%,南京特種氣體)至反應腔內,時間為2分鐘,以將在pt表面解離吸附的二茂鐵的有機配體部分通過氧化燃燒掉,並轉化為feox,從而實現feox在催化劑表面的沉積;(d)並用高純n2氣繼續吹掃5分鐘重複上述步驟(a-d)5次,即做5個原子層沉積周期。從反應腔內取出樣品,獲得pt-5cfeox/sio2催化劑。所獲催化劑中pt的質量含量仍是3.2wt%,幾乎不變,feox助劑以鐵元素計的質量含量是0.15wt%。
富氫氣氛中co優先氧化反應活性測試:反應氣中的co:o2:h2的體積比為1:0.5:48。催化劑:上述pt-5cfeox/sio2催化劑100mg與1g石英砂研磨均勻混合;反應器:u-型石英管;催化劑預處理:首先在10%o2/he氣氛中在200℃處理1小時,隨後切換為10%h2/he,繼續處理2小時,最後用1%co+0.5%o2+48%h2+50.5%he的反應氣體再處理20分鐘。反應氣體的組成:1%co+0.5%o2+48%h2+50.5%he,反應氣流速為60ml/min。在-80~200℃溫度區間內對催化劑進行活性測試,測試結果如圖4所示,從該圖1中可以看出,利用本發明製備的寬溫催化劑,能夠在-80~200℃溫度區間內實現富氫氣氛中co的高轉化率轉化。
實施例3-8:pt-3cfeox/sio2催化劑製備以及富氫氣氛中co優先氧化反應活性
按照實施例2的相同程序,只是分別利用二甲氨基二茂鐵(cpfec5h4chn(ch3)2,sigmaaldrich)、雙(2,4-二甲基戊二烯基)鐵(fe(2,4-c7h11)2,自合成)、(2,2,6,6-四甲基-3,5-庚二酮酸)鐵(iii)(fe(thd)3,alfaaesar)、雙(n,n′-二叔丁基乙脒基)鐵(stremchemicals)、羰基鐵(fe(co)5,sigmaaldrich)和叔丁醇鐵(fe2(otbu)6,sigmaaldrich)代替二茂鐵(fecp2,sigmaaldrich)作為feox助劑的金屬前驅體,並且沉積周期為3個,獲得相應的六個pt-3cfeox/sio2催化劑樣品。在所獲得的這六 個催化劑樣品中,pt的質量含量為3.2wt%,feox助劑以鐵元素計的質量含量分別為0.18wt%、0.14wt%、0.15wt%、0.2wt%、0.3wt%和0.26wt%。
將以上獲得的各個催化劑,在實施例2中的相同程序和條件下,在富氫氣氛中co優先氧化反應活性測試中均表現出了與實施例2類似的在寬溫度範圍內實現富氫氣氛中co的高轉化率轉化。
實施例9:pt-1cfeox/c催化劑製備以及富氫氣氛中co優先氧化反應活性
pt/c催化劑前驅體的獲得:該pt/c催化劑前驅體是商用催化劑,購於sigmaaldrich,pt在催化劑中的質量含量為5wt%,根據高分辨電鏡照片,pt顆粒的尺寸為2.1±0.3nm。
feox助劑的沉積:通過電阻加熱,把粘滯流動型原子層沉積反應設備(arradiance)的反應腔溫度加熱到120℃,使用的feox助劑的金屬前驅體是二茂鐵(fecp2,sigmaaldrich),利用加熱套,把該金屬前驅體源容器的溫度加熱到90℃,以獲得足夠的二茂鐵前驅體蒸汽壓。將200mgpt/c商用催化劑前驅體樣品放置到原子層沉積反應腔內。打開二茂鐵容器和原子層沉積反應腔之間的隔離閥門,二茂鐵蒸汽混入高純n2(99.999%,南京特種氣體),並被高純n2引入到原子層沉積反應腔內。二茂鐵並在pt/c催化劑前驅體中的pt顆粒表面發生解離吸附,繼而實現fe在pt表面的成核生成,引入時間為2.5分鐘。關閉二茂鐵源後,引入作為氧化劑的高純o2(99.999%,南京特種氣體)至反應腔內,時間為3分鐘,以將在pt表面解離吸附的二茂鐵的有機配體部分通過氧化燃燒掉,並轉化為feox,從而實現feox在催化劑表面的沉積。最後,從反應腔內取出樣品,獲得pt-1cfeox/c催化劑。所獲催化劑中feox助劑的鐵元素的質量含量是8.6wt%。
富氫氣氛中co優先氧化反應活性測試:反應氣中的co:o2:h2的體積比為1:1:48。催化劑:上述pt-1cfeox/c催化劑100mg與1g石英砂研磨均勻混合;反應器:u-型石英管;催化劑預處理:首先在10%o2/he氣氛中在200℃處理1小時,隨後切換為10%h2/he,繼續處理2小時,最後 用1%co+1%o2+48%h2+50%he的反應氣體再處理20分鐘。反應氣體的組成:1%co+1%o2+48%h2+50%he,反應氣流速為60ml/min。在-80~200℃溫度區間內對催化劑進行活性測試,測試結果如圖5所示,利用本發明製備的寬溫催化劑,能夠在-50~120℃溫度區間實現co的完全轉化。
實施例10:pt-1cfeox/tio2催化劑製備以及富氫氣氛中co優先氧化反應活性
pt/tio2催化劑前驅體製備:該催化劑前驅體是利用原子層沉積的辦法獲取的。使用粘滯流動型原子層沉積反應器(arradiance),原子層沉積的溫度為250℃,實驗的pt前驅體為(三甲基)甲基環戊二烯合鉑(iv)(mecpptme3,stremchemicals),利用加熱套,把該金屬前驅體源容器的溫度加熱到70℃,以獲得足夠的mecpptme3前驅體蒸汽壓。氧化劑為高純o2(99.999%,南京特種氣體),惰性氣體為高純n2(99.999%,南京特種氣體)。在催化劑製備過程中,把700mgtio2載體(50m2/g,degussa)放入其反應腔內,(a)利用高純n2把mecpptme3的蒸氣引入到反應腔內,時間為4分鐘;(b)把mecpptme3源停止後,用高純n2氣吹掃5分鐘;(c)引高純o2進入反應腔內,時間為2分鐘;(d)然後再關掉o2源,並用高純n2氣吹掃5分鐘。重複上述步驟(a-d)2次,即做2個原子層沉積周期。從反應腔內取出樣品,獲得pt/sio2催化劑前驅體。其中pt的質量含量為3.7wt%,根據高分辨電鏡結果,pt顆粒尺寸約為3nm。
feox助劑的沉積:通過電阻加熱,把粘滯流動型原子層沉積反應設備(arradiance)的反應腔溫度加熱到120℃,使用的feox助劑的金屬前驅體是二茂鐵(fecp2,sigmaaldrich),利用加熱套,把該金屬前驅體源容器的溫度加熱到90℃,以獲得足夠的二茂鐵前驅體蒸汽壓。將上述200mgpt/tio2催化劑前驅體樣品放置到原子層沉積反應腔內。打開二茂鐵源容器和原子層沉積反應腔之間的隔離閥門,二茂鐵蒸汽混入高純n2(99.999%,南京特種氣體),並被高純n2引入到原子層沉積反應腔內。二茂鐵並在pt/tio2催化劑前驅體中的pt顆粒表面發生解離吸附,繼而實現fe在pt表面的成核生成,引入時間為2.5分鐘。關閉二茂鐵源後,引入作為氧化 劑的高純o2(99.999%,南京特種氣體)至反應腔內,時間為3分鐘,以將在pt表面解離吸附的二茂鐵的有機配體部分通過氧化燃燒掉,並轉化為feox,從而實現feox在催化劑表面的沉積。從反應腔內取出樣品,獲得pt-1cfeox/tio2催化劑。所獲催化劑中pt的質量含量為3.7wt%,幾乎保持不變,fe的質量含量是0.17wt%。
富氫氣氛中co優先氧化反應活性測試:反應氣中的co:o2:h2的體積比為1:1:48。催化劑:上述pt-1cfeox/tio2催化劑100mg與1g石英砂研磨均勻混合;反應器:u-型石英管;催化劑預處理:首先在10%o2/he氣氛中在200℃處理1小時,隨後切換為10%h2/he,繼續處理2小時,最後用1%co+1%o2+48%h2+50%he的反應氣體再處理20分鐘。反應氣體的組成:1%co+1%o2+48%h2+50%he,反應氣流速為60ml/min。在-80~200℃溫度區間內對催化劑進行活性測試,測試結果如圖6所示,利用本發明製備的寬溫催化劑,能夠在-70~100℃溫度區間實現co的完全轉化。
實施例11:pt-1cfeox/al2o3催化劑製備以及富氫氣氛中co優先氧化反應活性
pt/al2o3催化劑前驅體的製備:利用原子層沉積的辦法獲取的。使用粘滯流動型原子層沉積反應器(arradiance),原子層沉積的溫度為250℃,實驗的pt前驅體為(三甲基)甲基環戊二烯合鉑(iv)(mecpptme3,stremchemicals),利用加熱套,把該金屬前驅體源容器的溫度加熱到70℃,以獲得足夠的mecpptme3前驅體蒸汽壓。氧化劑為高純o2(99.999%,南京特種氣體),惰性氣體為高純n2(99.999%,南京特種氣體)。在催化劑製備過程中,把400mgal2o3載體(50m2/g,alfaaesar)放入其反應腔內,(a)利用高純n2把mecpptme3的蒸氣引入到反應腔內,時間為3分鐘;(b)把mecpptme3源停止後,用高純n2氣吹掃5分鐘;(c)引高純o2進入反應腔內,時間為2分鐘;(d)然後再關掉o2源,並用高純n2氣吹掃5分鐘。重複上述步驟(a-d)2次,即做2個原子層沉積周期。從反應腔內取出樣品,獲得pt/sio2催化劑前驅體。其中pt的質量含量為~3.7wt%, 根據高分辨電鏡結果,pt顆粒尺寸為~3nm。
feox助劑的沉積:通過電阻加熱,把粘滯流動型原子層沉積反應設備(arradiance)的反應腔溫度加熱到120℃,使用的feox助劑的金屬前驅體是二茂鐵(fecp2,sigmaaldrich),利用加熱套,把該金屬前驅體源容器的溫度加熱到90℃,以獲得足夠的二茂鐵前驅體蒸汽壓。將上述200mgpt/al2o3催化劑前驅體樣品放置到原子層沉積反應腔內。打開二茂鐵源容器和原子層沉積反應腔之間的隔離閥門,二茂鐵蒸汽混入高純n2(99.999%,南京特種氣體),並被高純n2引入到原子層沉積反應腔內。二茂鐵並在pt/al2o3催化劑前驅體中的pt顆粒表面發生解離吸附,繼而實現fe在pt表面的成核生成,引入時間為2.5分鐘。關閉二茂鐵源後,引入作為氧化劑的高純o2(99.999%,南京特種氣體)至反應腔內,時間為3分鐘,以將在pt表面解離吸附的二茂鐵的有機配體部分通過氧化燃燒掉,並轉化為feox,從而實現feox在催化劑表面的沉積。從反應腔內取出樣品,獲得pt-1cfeox/al2o3催化劑。所獲催化劑中pt的質量含量為~3.7wt%,幾乎保持不變,fe的質量含量是0.15wt%。
富氫氣氛中co優先氧化反應活性測試:反應氣中的co:o2:h2的體積比為1:1:48。催化劑:上述pt-1cfeox/al2o3催化劑100mg與1g石英砂研磨均勻混合;反應器:u-型石英管;催化劑預處理:首先在10%o2/he氣氛中在200℃處理1小時,隨後切換為10%h2/he,繼續處理2小時,最後用1%co+0.5%o2+48%h2+50.5%he的反應氣體再處理20分鐘。反應氣體的組成:1%co+1%o2+48%h2+50.5%he,反應氣流速為60ml/min。在-80~200℃溫度區間內對催化劑進行活性測試,測試結果如圖7所示,利用本發明製備的寬溫催化劑,能夠在50-200℃溫度區間實現co的完全轉化。
實施例12:pt-1ccoox/sio2催化劑製備以及富氫氣氛中co優先氧化反應活性
pt/sio2催化劑前驅體製備:首先以矽酸四乙酯(teos)為原料,通過鹼性水解的方法合成單分散二氧化矽小球(sio2)。其次將合成的二氧化矽 小球進行300℃的高溫煅燒處理。此後,用3-氨基丙基-三乙氧基矽烷(atpes)對煅燒後的二氧化矽小球表面進行修飾(atpes-sio2)。將1.4g的atpes-sio2和7.9ml的氯鉑酸加入30ml乙醇中,室溫下攪拌24小時,離心烘乾,得pt/sio2。其中催化劑中pt的質量含量是3.2wt%,pt顆粒尺寸小於1.5nm。
coox助劑的沉積:通過電阻加熱,把粘滯流動型原子層沉積反應設備(arradiance)的反應腔溫度加熱到150℃,使用的coox助劑的金屬前驅體是二茂鈷(cocp2,sigmaaldrich),利用加熱套,把該金屬前驅體源容器的溫度加熱到90℃,以獲得足夠的二茂鈷前驅體蒸汽壓。將上述200mgpt/sio2催化劑前驅體樣品放置到原子層沉積反應腔內。打開二茂鈷源容器和原子層沉積反應腔之間的隔離閥門,二茂鈷蒸汽混入高純n2(99.999%,南京特種氣體),並被高純n2引入到原子層沉積反應腔內。二茂鈷並在pt/sio2催化劑前驅體中的氯鉑酸根離子表面發生化學吸附,繼而實現co在pt離子上的成核生成,引入時間為3分鐘。關閉二茂鈷源後,引入作為氧化劑的高純o2(99.999%,南京特種氣體)至反應腔內,時間為3分鐘,以將在pt表面解離吸附的二茂鈷的有機配體部分通過氧化燃燒掉,並轉化為coox,從而實現coox在催化劑表面的沉積。最後,從反應腔內取出樣品,獲得pt-1ccoox/sio2催化劑。所獲催化劑中pt的質量含量是3.2wt%,幾乎保持不變,coox助劑以co元素計的質量含量是0.9wt%。
富氫氣氛co優先氧化活性測試:反應氣中的co:o2:h2的體積比為1:0.5:48。催化劑:[080]中所述pt-1cfeox/sio2(wi-u)催化劑100mg與1g石英砂研磨均勻混合;反應器:u-型石英管;催化劑預處理:首先在10%o2/he氣氛中在250℃處理2小時,隨後切換為10%h2/he,繼續處理1小時,最後用1%co+0.5%o2+48%h2+50.5%he的反應氣體再處理20分鐘。反應氣體的組成:1%co+0.5%o2+48%h2+50.5%he,反應氣流速為60ml/min。在30~200℃溫度區間內對催化劑進行活性測試,測試結果如圖8所示,利用本發明製備的寬溫催化劑,能夠在30~140℃溫度區間實現co的完全轉化。
實施例13-18:pt-1ccoox/sio2催化劑製備以及富氫氣氛中co優先氧化反應活性
按照實施例12的相同程序,只是分別利用乙醯丙酮鈷、雙(n,n′-二異丙基乙脒基)鈷、二羰基環戊二烯鈷、叔丁基三羰基鈷、(2,2,6,6-四甲基-3,5-庚二酮酸)鈷和2-甲氧基乙醇鈷(均購自sigmaaldrich)代替二茂鈷(cocp2,sigmaaldrich)作為coox助劑的金屬前驅體,並且沉積周期為1個,獲得相應的六個pt-1ccoox/sio2催化劑樣品。在所獲得的這六個催化劑樣品中,pt的質量含量為3.2wt%,coox助劑以鈷元素計的質量含量分別為0.8wt%、0.7wt%、0.7wt%、0.8wt%、08wt%和0.9wt%。
將以上獲得的各個催化劑,在實施例12中的相同程序和條件下,在富氫氣氛中co優先氧化反應活性測試中均表現出了與實施例12類似的在寬溫度範圍內實現富氫氣氛中co的高轉化率轉化。
實施例19:pt-1cniox/sio2催化劑製備以及富氫氣氛co優先氧化活性
pt/sio2催化劑前驅體的製備:利用原子層沉積的辦法獲取的。使用粘滯流動型原子層沉積反應器(arradiance),原子層沉積的溫度為250℃,實驗的pt前驅體為(三甲基)甲基環戊二烯合鉑(iv)(mecpptme3,stremchemicals),利用加熱套,把該金屬前驅體源容器的溫度加熱到70℃,以獲得足夠的mecpptme3前驅體蒸汽壓。氧化劑為高純o2(99.999%,南京特種氣體),惰性氣體為高純n2(99.999%,南京特種氣體)。在催化劑製備過程中,把700mgsio2載體(300m2/g,alfaaesar)放入其反應腔內,(a)利用高純n2把mecpptme3的蒸氣引入到反應腔內,時間為5分鐘;(b)把mecpptme3源停止後,用高純n2氣吹掃10分鐘;(c)引高純o2進入反應腔內,時間為3分鐘;(d)然後再關掉o2源,並用高純n2氣吹掃5分鐘。重複上述步驟(a-d)2次,即做2個原子層沉積周期。從反應腔內取出樣品,獲得pt/sio2催化劑前驅體。其中pt的質量含量為3.6wt%,根據高分辨電鏡結果,pt顆粒尺寸為2.7±0.4nm。
niox助劑的沉積:通過電阻加熱,把粘滯流動型原子層沉積反應設備(arradiance)的反應腔溫度加熱到120℃,使用的niox助劑的金屬前驅體是二茂鎳(nicp2,sigmaaldrich),利用加熱套,把該金屬前驅體源容器 的溫度加熱到90℃,以獲得足夠的二茂鎳前驅體蒸汽壓。將上述150mgpt/sio2催化劑前驅體樣品放置到原子層沉積反應腔內。打開二茂鎳源容器和原子層沉積反應腔之間的隔離閥門,二茂鎳蒸汽混入高純n2(99.999%,南京特種氣體),並被高純n2引入到原子層沉積反應腔內。二茂鎳在pt/sio2催化劑前驅體中的pt納米顆粒表面發生解離吸附,繼而實現ni在pt顆粒上的成核生成,引入時間為2.5分鐘。關閉二茂鎳源後,引入作為氧化劑的高純o2(99.999%,南京特種氣體)至反應腔內,時間為2分鐘,以將在pt表面解離吸附的二茂鎳的有機配體部分通過氧化燃燒掉,並轉化為niox,從而實現niox在催化劑表面的沉積。最後,從反應腔內取出樣品,獲得pt-1cniox/sio2催化劑。所獲催化劑中pt的質量含量是3.6wt%,幾乎保持不變,niox助劑以ni元素計的質量含量是1.23wt%。
富氫氣氛中co優先氧化反應活性測試:反應氣中的co:o2:h2的體積比為1:0.5:48。催化劑:上述pt-1cniox/sio2催化劑100mg與1g石英砂研磨均勻混合;反應器:u-型石英管;催化劑預處理:首先在10%o2/he氣氛中在200℃處理2小時,隨後切換為10%h2/he,繼續處理1小時,1%co+0.5%o2+48%h2+50.5%he,反應氣流速為60ml/min。在-80~200℃溫度區間內對催化劑進行活性測試,測試結果如圖9所示利用本發明製備的寬溫催化劑,能夠在35~60℃溫度區間實現co的完全轉化。
實施例20-24:pt-1cniox/sio2催化劑製備以及富氫氣氛中co優先氧化反應活性
按照實施例19的相同程序,只是分別利用乙醯丙酮鎳、雙(n,n′-二異丙基乙脒基)鎳、(2,2,6,6-四甲基-3,5-庚二酮酸)鎳(ii)、二丁基二硫代氨基甲酸鎳(ii)和2-甲氧基乙醇鎳(均購自sigmaaldrich)代替二茂鎳(nicp2,sigmaaldrich)作為niox助劑的金屬前驅體,並且沉積周期為1個,獲得相應的六個pt-3cniox/sio2催化劑樣品。在所獲得的這五個催化劑樣品中,pt元素的質量含量為3.6wt%,niox助劑以鎳元素計的質量含量分別為1.0wt%、0.8wt%、0.7wt%、0.7wt%、0.7wt%和0.7wt%。
將以上獲得的各個催化劑,在實施例19中的相同程序和條件下,在 富氫氣氛中co優先氧化反應活性測試中均表現出了與實施例19類似的在寬溫度範圍內實現富氫氣氛中co的高轉化率轉化。
實施例25:ir-2cfeox/sio2催化劑製備以及富氫氣氛中co優先氧化反應活性
ir/sio2催化劑前驅體製備:ir/sio2催化劑前驅體是用浸漬法合成獲得的。把480mgsio2加入含有2.12ml氯銥酸(2.46×10-3m)的水溶液中,攪拌24小時。再在80℃的溫度下蒸乾水溶液,然後放入70℃烘箱烘乾,得到ir/sio2催化劑。催化劑中ir的質量含量是3.7wt%,ir顆粒尺寸為1.50±0.6nm。
feox助劑的沉積:通過電阻加熱,把粘滯流動型原子層沉積反應設備(arradiance)的反應腔溫度加熱到200℃,使用的feox助劑的金屬前驅體是二茂鐵(fecp2,sigmaaldrich),利用加熱套,把該金屬前驅體源容器的溫度加熱到90℃,以獲得足夠的二茂鐵前驅體蒸汽壓。將上述200mgir/sio2催化劑前驅體樣品放置到原子層沉積反應腔內。(a)打開二茂鐵容器和原子層沉積反應腔之間的隔離閥門,二茂鐵蒸汽混入高純n2(99.999%,南京特種氣體),並被高純n2引入到原子層沉積反應腔內。二茂鐵並在ir/sio2催化劑前驅體中的ir顆粒表面發生解離吸附,繼而實現fe在ir表面的成核生成,引入時間為2.5分鐘。(b)關閉二茂鐵源後,並用高純n2氣繼續吹掃5分鐘,(c)引入作為氧化劑的高純o2(99.999%,南京特種氣體)至反應腔內,時間為2.5分鐘,以將在ir表面解離吸附的二茂鐵的有機配體部分通過氧化燃燒掉,並轉化為feox,從而實現feox在催化劑表面的沉積;(d)並用高純n2氣繼續吹掃5分鐘重複上述步驟(a-d)2次,即做2個原子層沉積周期。從反應腔內取出樣品,獲得ir-2cfeox/sio2催化劑。所獲催化劑中ir的質量含量是3.7wt%,幾乎保持不變,feox助劑中fe元素的質量含量是0.12wt%。
富氫氣氛中co優先氧化反應活性測試:反應氣中的co:o2:h2的體積比為1:1:48。催化劑:[087]中所述ir-1cfeox/sio2催化劑100mg與1g石英砂研磨均勻混合;反應器:u-型石英管;催化劑預處理:首先在10%o2/ar 氣氛中在200℃處理1小時,隨後切換為10%h2/ar,繼續處理2小時。反應氣體的組成:1%co+1%o2+48%h2+50%ar,反應氣流速為60ml/min。在室溫~200℃溫度區間內對催化劑進行活性測試,測試結果如圖10所示,利用本發明製備的寬溫催化劑,能夠在60~180℃溫度區間實現co的完全轉化。
穩定性測試:在上述ir-1cfeox/sio2催化劑的富氫氣氛中co優先氧化反應的穩定性測試,反應氣中的co:o2:h2的體積比為1:1:48。催化劑的用量、預處理以及反應條件如上所述。把反應溫度保持在80℃維持不變,對樣品連續測試20小時。把反應溫度為80℃。穩定性測試時間為20小時。如圖11所示,該催化劑能夠在80℃保持20小時而無任何催化劑失活現象。
實施例26:ir-1ccoox/sio2催化劑製備以及富氫氣氛中co優先氧化反應活性
ir/sio2催化劑前驅體製備:首先以矽酸四乙酯(teos)為原料,通過鹼性水解的方法合成單分散二氧化矽小球(sio2)。其次將合成的二氧化矽小球進行300℃的高溫煅燒處理。此後,用3-氨基丙基-三乙氧基矽烷(atpes)對煅燒後的二氧化矽小球表面進行修飾(atpes-sio2)。將480mgatpes-sio2和2.12ml氯銥酸加入50ml水溶液中,室溫下攪拌24小時,再在80℃的溫度下蒸乾水溶液,烘乾,得到ir/sio2。
coox助劑的沉積:通過電阻加熱,把粘滯流動型原子層沉積反應設備(arradiance)的反應腔溫度加熱到150℃,使用的coox助劑的金屬前驅體是二茂鈷(cocp2,sigmaaldrich),利用加熱套,把該金屬前驅體源容器的溫度加熱到90℃,以獲得足夠的二茂鈷前驅體蒸汽壓。將上述200mgir/sio2催化劑前驅體樣品放置到原子層沉積反應腔內。打開二茂鈷源容器和原子層沉積反應腔之間的隔離閥門,二茂鈷蒸汽混入高純n2(99.999%,南京特種氣體),並被高純n2引入到原子層沉積反應腔內。二茂鈷並在ir/sio2催化劑前驅體中的氯銥酸根離子表面發生化學吸附,繼而實現co在pt離子上的成核生成,引入時間為3分鐘。關閉二茂鈷源後, 引入作為氧化劑的高純o2(99.999%,南京特種氣體)至反應腔內,時間為3分鐘,以將在ir表面解離吸附的二茂鈷的有機配體部分通過氧化燃燒掉,並轉化為coox,從而實現coox在催化劑表面的沉積。最後,從反應腔內取出樣品,獲得ir-1ccoox/sio2催化劑。所獲催化劑中co的質量含量是1.23wt%。
富氫氣氛中co優先氧化反應活性測試:反應氣中的co:o2:h2的體積比為1:1:48。催化劑:上述ir-1ccoox/sio2催化劑100mg與1g石英砂研磨均勻混合;反應器:u-型石英管;催化劑預處理:首先在10%o2/ar氣氛中在500℃處理1小時,隨後降溫到250℃,並切換為10%h2/he繼續處理1小時。反應氣體的組成:1%co+1%o2+48%h2+50%ar,反應氣流速為30ml/min。在20~200℃溫度區間內對催化劑進行活性測試,測試結果如圖12所示,利用本發明製備的寬溫催化劑,能夠在80~120℃溫度區間實現co的完全轉化。
實施例27:pt-1cfeox/sio2(fe(acac)3)催化劑製備以及富氫氣氛中co優先氧化反應活性
pt/sio2催化劑前驅體的製備:利用原子層沉積的辦法獲取的。使用粘滯流動型原子層沉積反應器(arradiance),原子層沉積的溫度為250℃,實驗的pt前驅體為(三甲基)甲基環戊二烯合鉑(iv)(mecpptme3,stremchemicals),利用加熱套,把該金屬前驅體源容器的溫度加熱到70℃,以獲得足夠的mecpptme3前驅體蒸汽壓。氧化劑為高純o2(99.999%,南京特種氣體),惰性氣體為高純n2(99.999%,南京特種氣體)。在催化劑製備過程中,把700mgsio2載體(300m2/g,alfaaesar)放入其反應腔內,(a)利用高純n2把mecpptme3的蒸氣引入到反應腔內,時間為10分鐘;(b)把mecpptme3源停止後,用高純n2氣吹掃5分鐘;(c)引高純o2進入反應腔內,時間為3分鐘;(d)然後再關掉o2源,並用高純n2氣吹掃5分鐘。重複上述步驟(a-d)2次,即做2個原子層沉積周期。從反應腔內取出樣品,獲得pt/sio2催化劑前驅體。其中pt的質量含量為3.6wt%,根據高分辨電鏡結果,pt顆粒尺寸為2.7±0.4nm。
feox助劑的沉積:通過電阻加熱,把粘滯流動型原子層沉積反應設備(arradiance)的反應腔溫度加熱到120℃,使用的feox助劑的金屬前驅體是乙醯丙酮鐵(fe(acac)3,sigmaaldrich),利用加熱套,把該金屬前驅體源容器的溫度加熱到90℃,以獲得足夠的乙醯丙酮鐵前驅體蒸汽壓。將上述200mgpt/sio2催化劑前驅體樣品放置到原子層沉積反應腔內。打開乙醯丙酮鐵源容器和原子層沉積反應腔之間的隔離閥門,乙醯丙酮鐵蒸汽混入高純n2(99.999%,南京特種氣體),並被高純n2引入到原子層沉積反應腔內。乙醯丙酮鐵並在pt/sio2催化劑前驅體中的pt顆粒表面發生解離吸附,繼而實現fe在pt表面的成核生成,引入時間為2.5分鐘。關閉二茂鐵源後,引入作為氧化劑的高純o2(99.999%,南京特種氣體)至反應腔內,時間為3分鐘,以將在pt表面解離吸附的乙醯丙酮鐵的有機配體部分通過氧化燃燒掉,並轉化為feox,從而實現feox在催化劑表面的沉積。最後,從反應腔內取出樣品,獲得pt-1cfeox/sio2(fe(acac)3)催化劑。所獲催化劑中pt的質量含量是3.6wt%,幾乎保持不變,feox助劑的fe元素的質量含量是0.9%。
富氫氣氛中co優先氧化反應活性測試:反應氣中的co:o2:h2的體積比為1:0.5:48。催化劑:把[095]中所述pt-1cfeox/sio2(fe(acac)3)催化劑100mg與1g石英砂研磨均勻混合;反應器:u-型石英管;催化劑預處理:首先在10%o2/he氣氛中在200℃處理1小時,隨後切換為10%h2/he,繼續處理2小時,最後用1%co+0.5%o2+48%h2+50.5%he的反應氣體再處理20分鐘。反應氣體的組成:1%co+0.5%o2+48%h2+50.5%he,反應氣流速為60ml/min。在-80~200℃溫度區間內對催化劑進行活性測試,測試結果如圖13所示,利用本發明製備的寬溫催化劑,能夠在-30~42℃溫度區間實現co的完全轉化。
儘管本發明的具體實施方式已經得到詳細的描述,本領域技術人員將會理解。根據已經公開的所有教導,可以對那些細節進行各種修改和替換,這些改變均在本發明的保護範圍之內。本發明的全部範圍由所附權利要求及其任何等同物給出。