一種InDel位點基因型分型的方法與流程
2023-09-16 07:55:40 2
本發明涉及分子生物學技術領域,尤其涉及一種InDel位點基因型分型的方法。
背景技術:
插入缺失(Insertion and Deletion,簡稱InDel)是指同源序列的比對中出現的至少1bp核苷酸的差異,這種差異稱為InDels;InDel在基因組中的含量僅次於單核苷酸多態性(SNP)標記,具有數量多、分布廣泛、變異豐富等特點。根據基因組中插入缺失位點,設計擴增這些插入缺失位點的PCR引物,產生的多態性位點即為插入缺失(InDel)標記。插入缺失(InDel)標記在系統發育推斷、遺傳診斷、藥物設計等方面具有重要的應用潛力。
目前,常用的InDel分型方法主要是測序法和晶片法,測序法和晶片法實驗操作繁瑣、效率較低並且依賴相關大型儀器平臺作為支撐,費用昂貴。
技術實現要素:
有鑑於此,本發明的目的在於提供一種成本低、操作簡單靈活、準確的基因型分型的方法。
為了實現上述發明目的,本發明提供以下技術方案:
本發明提供了一種InDel位點基因型分型的方法,包括以下步驟:
1)將目的基因DNA序列進行T載體克隆測序,得到目的基因克隆產物序列,將所述目的基因克隆產物序列進行多重比對,得到InDel位點;
2)根據所述InDel位點設計PCR擴增引物,所述PCR擴增引物包括正向引物和反向引物,所述正向引物的5'末端採用螢光基團修飾;
3)用所述PCR擴增引物對待測樣本全基因組DNA進行PCR擴增獲得擴增產物,用毛細管螢光凝膠電泳檢測擴增產物,若擴增產物中有兩條條帶則該InDel位點的基因型為雜合型ID;若有一條條帶且片段大小與雜合型ID中大的片段相同,則該InDel位點基因型為插入純合型II;若有一條條帶且片段大小與雜合型ID中小的片段相同,則表示該InDel位點為缺失純合型DD。
優選的,所述螢光基團為FAM或HEX。
優選的,所述多重比對採用MEGA軟體中的ClustalW模塊。
優選的,所述待測樣本為楊木樹種。
優選的,所述目的基因為尿苷二磷酸葡糖醛酸脫羧酶基因1。
優選的,所述尿苷二磷酸葡糖醛酸脫羧酶基因1上的InDel位點分別為InDel1、InDel2、InDel3、InDel4和InDel5;所述InDel1位於尿苷二磷酸葡糖醛酸脫羧酶基因1全長第401bp到409bp處,所述InDel2位於第1399bp到1401bp處,InDel3位於第1427bp到1438bp,InDel4位於第2094bp到2103bp處,InDel5位於第4143bp到4152bp。
優選的,根據InDel1設計的引物為正向引物SEQ ID NO.1和反向引物SEQ ID NO.2;根據InDel2和InDel3設計的引物為正向引物SEQ ID NO.3和反向引物SEQ ID NO.4;根據InDel4設計的引物為正向引物SEQ ID NO.5和反向引物SEQ ID NO.6;根據InDel5設計的引物為正向引物SEQ ID NO.7和反向引物SEQ ID NO.8。
優選的,步驟3)中所述PCR擴增的條件為:95℃預變性5min;95℃變性30s,53~57℃退火30s,72℃延伸30s,35個循環;最後72℃延伸5min。
優選的,所述PCR擴增用擴增體系包括以下組分:0.7μl的待測樣品全基因組DNA,0.15μl的0.8UTaq酶,0.3μl 0.2mM的dNTPs,0.9μl 25mM的MgCl2,1.25μl的10×PCR buffer,0.4μl 100nmolL-1的正向引物和0.4μl 100nmolL-1反向引物和8.4μl的ddH2O。
優選的,所述基因組DNA的濃度為5~20ng/μl。
本發明的有益效果:本發明提供的InDel位點基因型分型的方法通過多重比對獲得目的基因InDel位點,根據InDel位點設計PCR擴增引物,包括正向引物和反向引物,採用螢光基團修飾正向引物的5』端,然後對待測樣本DNA進行PCR擴增,用毛細管螢光凝膠電泳檢測擴增產物確定所述待測樣本中InDel位點的基因型。本發明對引物組中的正向引物5'末端採用螢光基團進行修飾,可顯著地保留螢光信號強度、減少InDel位點的非特異性擴增,提高了基因型檢測的準確度;同時極大地簡化了實驗步驟和配套條件,並顯著地降低了實驗成本。
進一步的,在適宜退火溫度和PCR反應條件下,進一步提高螢光信號強度,提高基因型檢測的準確度。
附圖說明
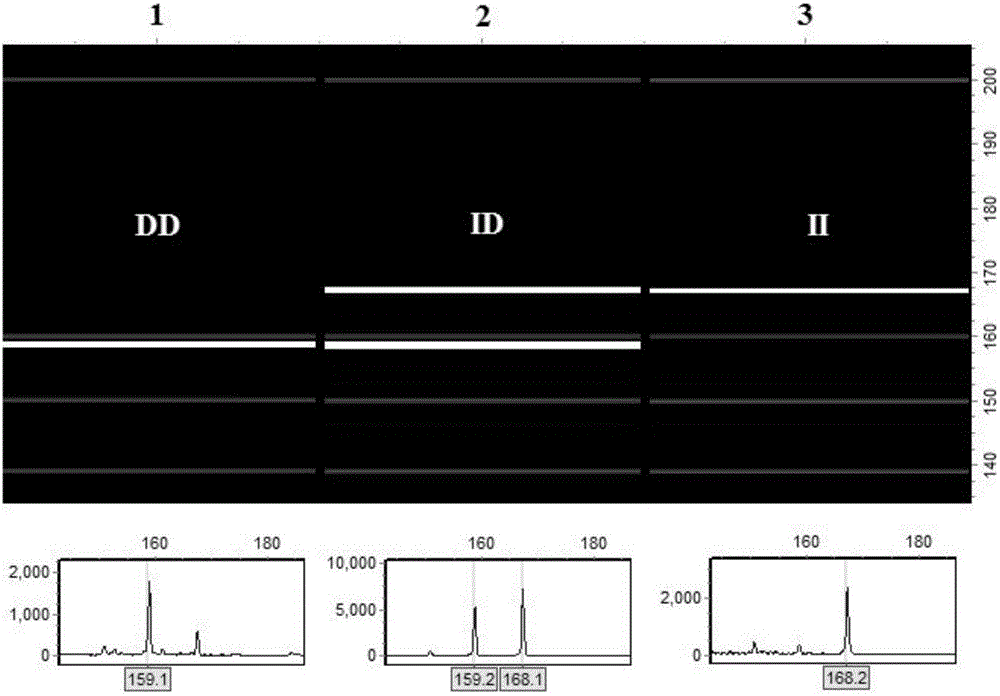
圖1為實施例1中毛細管螢光凝膠電泳結果圖。
具體實施方式
本發明提供了一種InDel位點基因型分型的方法,包括以下步驟:1)將目的基因DNA序列進行T載體克隆測序,得到目的基因克隆產物序列,將所述目的基因克隆產物序列進行多重比對,得到InDel位點;2)根據所述InDel位點設計PCR擴增引物,所述PCR擴增引物包括正向引物和反向引物,所述正向引物的5'末端採用螢光基團修飾;3)用所述PCR擴增引物對待測樣本DNA進行PCR擴增獲得擴增產物,毛細管螢光凝膠電泳檢測擴增產物,若擴增產物中有兩條目的條帶則該InDel位點的基因型為雜合型ID;若有一條目的條帶且片段大小與雜合型ID中大的片段相同,則該InDel位點基因型為插入純合型II;若有一條目的條帶且片段大小與雜合型ID中小的片段相同,則表示該InDel位點為缺失純合型DD。
本發明提供的InDel位點基因型分型的方法適用於林木樹種,優選的為楊木樹種,如本發明的實施例中所用毛白楊。
本發明將目的基因DNA序列進行T載體克隆測序,得到目的基因克隆產物序列。本發明中所述的目的基因為待測樣品的任一基因,優選的為尿苷二磷酸葡糖醛酸脫羧酶基因1,優選的所述尿苷二磷酸葡糖醛酸脫羧酶基因1的序列如SEQ ID NO.9所示。
在本發明選定目的基因後,將所述目的基因DNA序列進行T載體克隆測序,得到目的基因克隆產物序列。本發明對所述的T載體克隆測序的具體步驟沒有特殊限定,採用本領域常規的T載體轉化大腸桿菌DH52感受態細胞克隆測序方法步驟即可。
得到目的基因克隆產物序列後,本發明將所述目的基因克隆產物序列進行多重比對,得到InDel位點。在本發明中,所述的多重比對優選的採用MEGA軟體中的ClustalW模塊,具體的多重比對的參數優選的為軟體模塊中的默認參數。
具體的在本發明實施例中以尿苷二磷酸葡糖醛酸脫羧酶基因1為目的基因,對目的基因進行T載體克隆測序,後採用MEGA軟體中的ClustalW模塊進行多重比對,確定的InDel位點有5個,分別為InDel1、InDel2、InDel3、InDel4和InDel5;所述InDel1位於尿苷二磷酸葡糖醛酸脫羧酶基因1全長第401bp到409bp處,所述InDel2位於第1399bp到1401bp處,InDel3位於第1427bp到1438bp,InDel4位於第2094bp到2103bp處,InDel5位於第4143bp到4152bp。
本發明在獲得上述InDel位點後,分別根據上述InDel位點設計引物,包括正向引物和反向引物,所述正向引物的5'末端採用螢光基團修飾。優選的根據InDel1設計的引物為正向引物SEQ ID NO.1和反向引物SEQ ID NO.2;正向引物的序列為UXS1ID1F:5'-CTCCCGCCTTAACCCATCT-3';反向引物的序列為UXS1ID1R:5'-GTAACCACGATACGCAAACTC-3';優選的在本發明中針對InDel1設計的正向引物SEQ ID NO.1的5'末端採用螢光基團FAM進行修飾。
在本發明中,由於InDel2和InDel3在尿苷二磷酸葡糖醛酸脫羧酶基因1的位點相距較近,因此,設計同一對引物對上述兩個位點同時進行擴增;優選的根據InDel2和InDel3設計的引物為正向引物SEQ ID NO.3和反向引物SEQ ID NO.4;正向引物的序列為UXS1ID2F:5'-GTGGTTTTACTCGGTGCTTCT-3';反向引物的序列為UXS1ID2R:5'-GGCTCCAACTCAGTTCACACTT-3';優選的在本發明中針對InDel1設計的正向引物SEQ ID NO.3的5'末端採用螢光基團FAM進行修飾。
本發明中,根據InDel4設計的引物為正向引物SEQ ID NO.5和反向引物SEQ ID NO.6;正向引物的序列為UXS1ID4F:5'-TTCAGCACTCTACCAGTCCA-3';反向引物的序列為UXS1ID4R:5'-TCAGGCACACCACGTCAAA-3';優選的在本發明中針對InDel4設計的正向引物SEQ ID NO.5的5'末端採用螢光基團FAM進行修飾。
在本發明中,根據InDel5設計的引物為正向引物SEQ ID NO.7和反向引物SEQ ID NO.8;正向引物的序列為UXS1ID5F:5'-GGCTCTCTTCCGTAATCCA-3';反向引物的序列為UXS1ID5R:5'-CATTGCAAGGGAAAAACTACAC-3';優選的在本發明中針對InDel5設計的正向引物SEQ ID NO.7的5'末端採用螢光基團HEX進行修飾。
得到PCR擴增引物後,本發明用所述PCR擴增引物對待測樣本全基因組DNA進行PCR擴增獲得擴增產物,優選的,所述PCR擴增的條件為:95℃預變性5min;95℃變性30s,53~57℃退火30s,72℃延伸30s,35個循環;最後72℃延伸5min。所述退火溫度更優選的為55~56℃;所述PCR擴增的體系優選的包括以下組分:0.7μl的待測樣本全基因組DNA,0.15μl的0.8UTaq酶,0.3μl 0.2mM的dNTPs,0.9μl 25mM的MgCl2,1.25μl的10×PCR buffer,0.4μl 100nmolL-1的正向引物和0.4μl 100nmolL-1反向引物和8.4μl的ddH2O。
在本發明中所述待測樣本全基因組DNA採用本領域常規的基因組DNA的提取方法獲得,具體在本發明實施例中採用DNA提取試劑盒獲得,具體的可採用sigma公司的植物基因組DNA小量製備試劑盒;所述基因組DNA的濃度優選的為5~20ng/μl,更優選的為10~15ng/μl。
本發明在獲得擴增產物後,用毛細管螢光凝膠電泳檢測擴增產物的片段大小,本發明對所述的毛細管螢光凝膠電泳的方法和具體參數設置沒有特殊要求,採用本領域常規的方法和參數即可。所述毛細管螢光凝膠電泳檢測的原理如下:當擴增片段經過掃描儀CCD(charge-coupled device)掃描窗口時,螢光染料分子發出一定波長的螢光被CCD接收,CCD記錄每一個DNA片段通過的時間並以螢光峰值表示。理論上,螢光峰值出現的時間與擴增產物片段大小呈正相關,而峰值的高低與擴增產物的數量呈正相關。因此,在正向引物5'末端進行螢光基團修飾後,進行PCR擴增,通過檢測擴增產物螢光片段大小的差異,可以判斷目標區域靶位點的InDel類型。在本發明中,具體的,若擴增產物中有兩條條帶則該InDel位點的基因型為雜合型ID;若有一條條帶且片段大小與雜合型ID中大的片段相同,則該InDel位點基因型為插入純合型II;若有一條條帶且片段大小與雜合型ID中小的片段相同,則表示該InDel位點為缺失純合型DD。
下面結合實施例對本發明提供的InDel位點基因型分型的方法進行詳細的說明,但是不能把它們理解為對本發明保護範圍的限定。
實施例1
材料取自山東冠縣毛白楊基因庫(全國毛白楊種質資源庫)中435株個體。
提取每株個體的DNA後,按照地域分布,隨機挑選45株個體,逐一對其尿苷二磷酸葡糖醛酸脫羧酶基因1進行克隆測序,利用MEGA軟體中的ClustalW程序進行多重比對,確定候選InDel1位點。InDel1位於尿苷二磷酸葡糖醛酸脫羧酶基因1全長第401bp到409bp處,其所在區域的基因序列如:斜體為InDel1位點。
針對InDel1位點(黑體下劃線標註),整個自然群體包含兩種類型的DNA模板鏈,其中模板鏈I:模板鏈II:3'-GAGGGCGGAATTGGGTAGACTAGGTTGGTGGGTAAGAAGAGAGGGGAGAAGTTAAATGGTA---------NNNNNNNNNNNN-5'。設計合成一條5'末端用FAM螢光基團進行修飾的正向引物:SEQ ID NO.1為UXS1ID1F:5'-CTCCCGCCTTAACCCATCT-3',一條不進行螢光修飾的反向引物:SEQ ID NO.2為UXS1ID1R:5'-GTAACCACGATACGCAAACTC-3',擴增產物目的片段長度為168bp。
將已知InDel基因型為DD、ID和II的三個待測樣品的基因組DNA模板與所設計引物組合,進行PCR擴增,擴增程序為:預變性95℃5min,變性95℃30s,退火56℃30s,延伸72℃30s,35個循環,最後延伸72℃5min,4℃保存。12.5μl反應體系中包括:0.7μl 15ng/μl基因組DNA,0.15μl 0.8UTaq酶,0.3μl 0.2mM dNTPs,0.9μl 25mmolL-1MgCl2,1.25μl 10×PCRbuffer以及0.4μl100nmolL-1正向螢光標記引物和0.4μl 100nmolL-1反向引物和8.4μl的ddH2O。將PCR擴增的產物進行毛細管螢光凝膠電泳檢測,每條泳道檢測一株個體的基因型,根據檢測結果確定待測樣品的基因型。結果如圖1所示,圖1中上方為毛細管螢光電泳成像圖,下部為螢光峰值圖。其中,三個泳道內,字母DD表示該個體InDel1位點的基因型為deletion/deletion;字母ID表示該個體InDel1位點的基因型為insertion/deletion;字母II表示該個體InDel1位點的基因型為insertion/insertion。深灰色亮帶為核苷酸分子量Marker,最右側數值為核苷酸分子量大小。根據目的片段的大小,可以確定三株個體的基因型分別是DD、ID和II。
然後再取自山東冠縣毛白楊基因庫中435株個體中按照上述方法進行InDel1位點的基因型檢測,其中有27株個體基因型為DD,有383株個體基因型為ID,有25株個體基因型為II。
實施例2
材料取自山東冠縣毛白楊基因庫(全國毛白楊種質資源庫)中435株個體。
提取每株個體的DNA後,按照地域分布,隨機挑選45株個體,逐一對其尿苷二磷酸葡糖醛酸脫羧酶基因1進行克隆測序,利用MEGA軟體中的ClustalW程序進行多重比對,確定候選InDel2和InDel3位點。InDel2位於尿苷二磷酸葡糖醛酸脫羧酶基因1全長第1399bp到1401bp處,InDel3位點位於第1427bp到1438bp處,其所在區域的基因序列如下:斜體為InDel2位點;斜體為InDel3位點)進行基因型檢測。
設計合成一條5'末端用FAM螢光基團進行修飾的正向引物:SEQ ID NO.3為UXS1ID2F:5'-GTGGTTTTACTCGGTGCTTCT-3',一條不進行螢光修飾的反向引物:SEQ ID NO.4為UXS1ID2R:5'-GGCTCCAACTCAGTTCACACTT-3',擴增產物目的片段長度為209bp。
按照實施例1的PCR擴增程序和體系對山東冠縣毛白楊基因庫中435株個體尿苷二磷酸葡糖醛酸脫羧酶基因1中的InDel2和InDel3位點基因型進行檢測,檢測結果為:有373株個體基因型為ID,有62株個體基因型為II;InDel3位點基因型檢測結果為:435株個體基因型均為ID。
實施例3
材料取自山東冠縣毛白楊基因庫(全國毛白楊種質資源庫)中435株個體。
提取每株個體的DNA後,按照地域分布,隨機挑選45株個體,逐一對其尿苷二磷酸葡糖醛酸脫羧酶基因1進行克隆測序,利用MEGA軟體中的ClustalW程序進行多重比對,確定候選InDel4和InDel5位點。InDel4位於尿苷二磷酸葡糖醛酸脫羧酶基因1全長第2094bp到2103bp處,其所在區域基因序列如下:斜體為InDel4位點;InDel5位於尿苷二磷酸葡糖醛酸脫羧酶基因1全長第4143bp到4152bp處,其所在區域的基因序列如下:斜體為InDel5位點,同時進行多位點基因型檢測。
根據待檢測位點InDel4和DNA模板鏈的序列特點,設計合成一條5'末端用FAM螢光基團進行修飾的正向引物,SEQ ID NO.5為UXS1ID4F:5'-TTCAGCACTCTACCAGTCCA-3'和一條不進行螢光修飾的反向引物SEQ ID NO.6為UXS1ID4R:5'-TCAGGCACACCACGTCAAA-3',擴增產物目的片段長度為160bp。
根據待檢測位點InDel5和DNA模板鏈的序列特點,設計合成一條5'末端用HEX螢光基團進行修飾的正向引物:SEQ ID NO.7為UXS1ID5F:5'-GGCTCTCTTCCGTAATCCA-3'和一條不進行螢光修飾的反向引物SEQ ID NO.8為UXS1ID5R:5'-CATTGCAAGGGAAAAACTACAC-3',擴增產物目的片段長度為125bp。
將待測樣品的基因組DNA模板與上述SEQ ID NO.5~SEQ ID NO.7引物組合,進行PCR擴增,擴增程序為:預變性95℃5min,變性95℃30s,退火57℃30s,延伸72℃30s,35個循環,最後延伸72℃5min,4℃保存。12.5μl反應體系中包括:0.7μl 15ng/μl基因組DNA,0.15μl 0.8UTaq酶,0.3μl 0.2mM dNTPs,0.9μl 25mmolL-1MgCl2,1.25μl 10×PCR buffer以及0.4μl 100nmolL-1正向螢光標記引物和0.4μl 100nmolL-1反向引物和8.4μl的ddH2O。將PCR擴增的產物進行毛細管螢光凝膠電泳檢測,每條泳道檢測一株個體的基因型,根據檢測結果確定待測樣品的基因型。對山東冠縣毛白楊基因庫中435株個體尿苷二磷酸葡糖醛酸脫羧酶基因1中的InDel4和InDel5位點基因型進行檢測,InDel4位點檢測結果為:有22株個體基因型為DD,有413株個體基因型為ID。InDel5位點基因型檢測結果為:有364株個體基因型為ID,有71株個體基因型為II。
由以上實施例可知,應用本發明所述的InDel位點基因型分型方法,對引物組中的正向引物5'末端採用螢光基團進行修飾,在最佳退火溫度和PCR反應條件下,可以顯著地保留螢光信號強度;利用毛細管螢光電泳,通過檢測螢光峰值出現的時間和峰值來判斷擴增產物片段大小和數量,進而判斷靶位點的InDel類型,實現對群體中所有個體的待測InDel位點進行基因型分型。應用毛細管螢光凝膠電泳技術檢測擴增產物大小,將檢測精度提高到1bp以內;同時,通過在正向引物5'末端修飾不同的螢光基團,可同時對多位點的基因型進行分型。因此不僅增加了檢測結果的精確性和可靠性,而且極大地簡化了實驗步驟和配套條件,並顯著地降低了實驗成本。
以上所述僅是本發明的優選實施方式,應當指出,對於本技術領域的普通技術人員來說,在不脫離本發明原理的前提下,還可以做出若干改進和潤飾,這些改進和潤飾也應視為本發明的保護範圍。