一種雙酚A‑雙(二苯基磷酸酯)的連續化製備方法與流程
2023-12-05 20:00:41 8
本發明屬於化工合成技術領域,具體涉及一種雙酚A-雙(二苯基磷酸酯)的連續化製備方法。
背景技術:
雙酚A-雙(二苯基磷酸酯)(簡稱BDP)分子式為C39H34O8P2,分子量為692,其結構式如下:
,
雙酚A-雙(二苯基磷酸酯)是無色或淡黃色液體,不溶於水,與二氯甲烷、甲苯、甲醇及甲基乙基酮等互相溶。密度(25℃)1.26g/㎝3,粘度(40℃)約2200mpa.s,TGA失重5%、10%及50%的溫度分別為371℃、397℃及444℃。
其為無滷型阻燃劑,廣泛用於PC/ABS,PPO/HIPS和其它聚合物。
目前,關於製備雙酚A-雙(二苯基磷酸酯)阻燃劑的方法,常用的雙酚A雙(二苯基磷酸酯)是在交聯反應釜中投入三氯氧磷、催化劑,再升溫至反應溫度,在回流條件下分批投入雙酚A,實現中間體製備,保溫一段時間後,再蒸出多餘的磷醯氯,與苯酚封端反應,製得粗品,再經酸洗、鹼洗、水洗、蒸餾、過濾製得BDP成品。經中國(專利檢索)得CN101456879A、200810139241.9、2000817989.1、1999816900.5、200380102119.X等,發現國內外大部分是以間歇合成反應或者部分連續生產來完成的。該種生產工藝優點是每套裝置獨立運行,獨立控制,互不幹擾。缺點是生產不能連續,反應條件不穩定,完成反應時間較長,達規模生產需要投入大量的設備、儀器、人員,且不能做到批產品質量的穩定,投資規模龐大。另外,根據市場需求進一步擴大產品規模,若採用大規模設備(列入10000L釜以上),可以提高單臺反應釜的產量,但由於反應釜的增大其傳質、傳熱效率更差,產品質量和收率更加難以達到工藝要求,而且反應釜越大其操作的危險性越大。為了保證產品質量收率,提高產量,現有工藝方法往往採用多臺反應釜和多套裝置進行,這樣大大增加了設備的投資和維護費用。
因此,提供一種連續化製備雙酚A-雙(二苯基磷酸酯)的方法,具有十分重要的意義。
技術實現要素:
為了克服現有技術中存在的上述問題,即雙酚A-雙(二苯基磷酸酯)間歇生產的缺點,本發明的目的在於提供一種雙酚A-雙(二苯基磷酸酯)的連續化製備方法及其裝置。它通過連續化工藝生產,達到安全、環保生產的目的,其批量生產產品穩定,適合大規模生產,且最大限度地降低了廢水排放,實現了循環經濟生產。
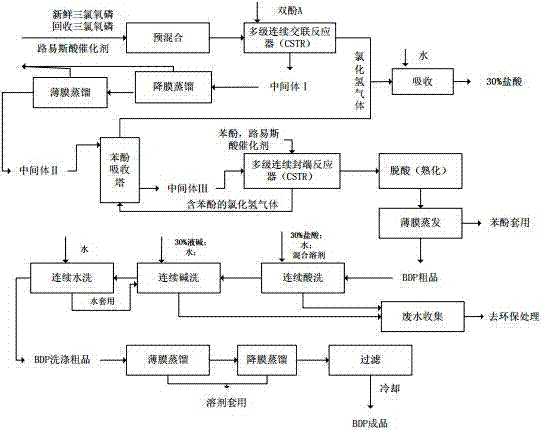
所述的一種雙酚A-雙(二苯基磷酸酯)的連續化製備方法,包括連續多級交聯反應工序、連續三氯氧磷回收工序、連續多級封端反應及苯酚回收工序、連續粗品精製工序、連續溶劑回收、過濾工序,其特徵在於包括如下步驟:
1)將三氯氧磷和路易斯酸催化劑預先混合,與雙酚A時分別連續加料到連續多級交聯反應器中,反應溫度控制在80-120℃,反應壓力為-0.015mpa-0.02mpa下進行交聯反應,得到中間體Ⅰ,即含有過量三氯氧磷的雙酚A磷醯氯縮聚物,反應產生的氯化氫經水吸收製成30%的副產物鹽酸;
2)將步驟1)得到的中間體Ⅰ連續蒸餾回收過量的三氯氧磷,得到中間體Ⅱ,即低三氯氧磷含量的雙酚A磷醯氯縮聚物;
3)將步驟2)得到的中間體Ⅱ經苯酚吸收塔吸收苯酚後得到中間體Ⅲ,即含少量苯酚的雙酚A磷醯氯縮聚物,中間體Ⅲ與苯酚分別連續加料到連續多級封端反應器中,在反應溫度90-160℃、反應壓力為-0.015mpa-0.02mpa下進行連續封端反應,得到含過量苯酚的雙酚A-雙(二苯基磷酸酯)粗品,粗品再經脫酸熟化、連續回收多餘的苯酚,得到雙酚A-雙(二苯基磷酸酯)粗品,反應產生的氯化氫經水吸收製成30%的副產物鹽酸,少量的苯酚一般是1-3%;
4)將步驟3)得到的雙酚A-雙(二苯基磷酸酯)粗品依次經連續酸洗、連續鹼洗、連續水洗,連續溶劑回收、過濾得到雙酚A-雙(二苯基磷酸酯)成品。
所述的一種雙酚A-雙(二苯基磷酸酯)的連續化製備方法,其特徵在於步驟1)中的路易斯酸催化劑包括無水氯化鎂、無水氯化鋁、氯化鋅、四氯化鈦,優選為無水氯化鎂,路易斯酸催化劑的投料重量為雙酚A重量的0.1%-2%。
所述的一種雙酚A-雙(二苯基磷酸酯)的連續化製備方法,其特徵在於步驟1)中的三氯氧磷為回收三氯氧磷和新三氯氧磷的混合物,回收三氯氧磷:新三氯氧磷:雙酚A的重量比為1.3-2.5:1.3-1.4:1。
所述的一種雙酚A-雙(二苯基磷酸酯)的連續化製備方法,其特徵在於步驟1)中連續多級交聯反應器包括四級反應器,第一級反應器的反應溫度95-100℃,第二級反應器的反應溫度105-110℃,第三級反應器的反應溫度110-120℃,第四級反應器的反應溫度110-120℃,物料在各反應器內的停留時間為1.5-2小時。
所述的一種雙酚A-雙(二苯基磷酸酯)的連續化製備方法,其特徵在於步驟2)中連續三氯氧磷回收工序採用兩級降膜蒸餾和兩級薄膜餾,最後一級薄膜蒸餾控制真空為150-400pa,物料溫度為120-130℃。
所述的一種雙酚A-雙(二苯基磷酸酯)的連續化製備方法,其特徵在於步驟3)中還能補加路易斯酸催化劑,中間體Ⅲ:苯酚:補加的路易斯酸催化劑的重量比為=1:0.78-0.086:0-0.004;粗品中的過量苯酚的重量含量為2-10%,優選為3-5%。
所述的一種雙酚A-雙(二苯基磷酸酯)的連續化製備方法,其特徵在於步驟3)中的連續多級封端反應器為四級反應器,第一級反應反應器的溫度100-105℃,第二級反應器的反應溫度120-130℃,第三級反應器的反應溫度140-150℃,第四級反應器的反應溫度140-150℃,各反應器的物料停留時間為1.5-2小時。
所述的一種雙酚A-雙(二苯基磷酸酯)的連續化製備方法,其特徵在於步驟3)中的苯酚回收採用一級或兩級薄膜蒸餾,蒸餾真空度為100-300pa,物料溫度為140-160℃,回收的苯酚用於套用。
所述的一種雙酚A-雙(二苯基磷酸酯)的連續化製備方法,其特徵在於步驟4)中連續粗品精製包括酸洗、鹼洗和水洗,連續酸洗工序中雙酚A-雙(二苯基磷酸酯)粗品:30%鹽酸:水:混合溶劑的重量配比為1:0.05-0.1:0.15-0.2:1.8-2.2,優選為1:0.07:0.17:2;連續鹼洗工序中酸洗粗品溶液、30%液鹼和水的重量配比為=1:0.02-0.026:0.1-0.14,優選為1:0.024:0.12;連續水洗工序中鹼洗粗品溶液與水的重量配比為為1:0.1-0.15,優選為1:0.12;連續酸洗、鹼洗和水洗中的洗滌溫度均為60-75℃;連續溶劑回收工序採用兩級降膜蒸餾和兩級或三級薄膜蒸餾,最後一級薄膜蒸餾真空度為30-100pa,優選為40-70pa,物料溫度為140-160℃,酸洗料層作為下一道工序的原料,分水器下層水排入廢水收集罐;下層水液排入廢水收集罐內,去三廢處理站處理;鹼洗料層作為下一道工序的原料,下層去廢水罐;下層水液排入廢水收集罐內,去三廢處理站處理;分水器下層去廢水罐;下層水液排入廢水收集罐內,水洗廢水配鹼洗液用,水洗過程中溫度控制在60-70℃。
所述的一種雙酚A-雙(二苯基磷酸酯)的連續化製備方法,其特徵在於物料輸送採用泵輸送和文丘裡管輸送,液體物料優選泵輸送,固體物料選擇文丘裡管輸送,輸送條件為真空或壓力輸送。
通過採用上述技術,與現有技術相比,本發明的有益效果如下:
1)本發明通過採用連續化工藝製備BDP,降低了生產成本,降低了操作人員勞動強度大幅,提高了勞動效率,且產品產量大幅提高,使批生產質量穩定,達到安全環保生產,其收率提升1-1.5%、色號提升至50以下、TPP控制在1%,比原有間歇裝置降低50%,符合高端客戶需求,且各產品質量指標波動小;
2)本發明通過採用先進的密閉投料方式,有效地解決原有工藝的固體投料困難及裝置氣密性問題,避免了反應物料潤溼進料管道,出現粘壁現象,隔絕空氣中的氧氣和水汽進入反應系統,進一步提升產品收率及質量;
3)本發明採用先進的塔式氣液分離、外循環設備有效的提高反應速率,加快反應進程,提高生產能力和裝置效率;採用連接苯酚回收技術使副產物鹽酸達到工業級水平,另外通過先進的回收技術,使苯酚及後續鹼量單耗減少,降低了成本,另外也進一步減輕環保壓力;
4)本發明連續反應過程中,回收的物料苯酚等全部套用於生產中,一方面降低成本、單耗,另一方面大大減輕環保壓力,提高了經濟效益,特別是由於苯酚是易揮發物質再和中間體反應過程中,會隨著氯化氫的溢出而夾帶。連續工藝採用中間體經苯酚洗滌塔去吸收夾帶的苯酚,一方面減少苯酚的單耗,另一方面提高副產物鹽酸的品質。
附圖說明
圖1為本發明的工藝流程圖。
具體實施方式
以下結合說明書附圖對本發明作進一步的描述,但本發明的保護範圍並不僅限於此:
實施例1 BDP的製備,其工藝流程如圖1所示,具體步驟如下:
1、連續多級交聯反應工序:
在預混釜內投入三氯氧磷2000Kg/h、回收三氯氧磷2500Kg/h,和氯化鎂15Kg/h,再將混合好的三氯氧磷,經泵增壓通過加熱至90-100℃,同時雙酚A1500kg/h連續按比例進料,在第一級交聯反應器進行交聯反應:反應物料經釜底經泵增壓上氣液分離器塔、冷凝器實現氣液分離,冷凝的三氯氧磷返回釜內。釜內反應溫度95-100℃,反應物料利用位差轉至後續反應器進行保溫,第二級交聯反應器反應溫度105-110℃,第三、四級交聯反應器反應溫度110-120℃,各反應器物料停留時間2小時。最後反應完的中間體Ⅰ放中間罐作為下一道工序的原料。
2、連續三氯氧磷回收工序
上述中間體Ⅰ經泵流量計計量以5500kg/h進料速度經過兩級降膜脫去大部分三氯氧磷,再經過兩級薄膜製得低三氯氧磷含量的中間體Ⅱ,最後一級工藝參數真空度300pa,蒸餾溫度120-125℃,蒸完氧磷的中間體Ⅱ放在暫存罐備用,中間體Ⅱ作為下一道洗滌工序的原料。
3、連續多級封端反應及苯酚回收工序:
上述蒸完三氯氧磷的中間體Ⅱ用泵經流量計以3000Kg/h的經進過苯酚洗滌塔,到中間體暫存罐,經泵增壓加熱至100-105℃,同時苯酚2350kg/h和氯化鎂10Kg/h連續按比例進料,再經靜態混合器混勻,送入第一級封端反應器:反應物料從反應釜體的底閥經泵增壓,再通過換熱器加熱至100℃,上氣液分離器塔、冷凝器,產生的氯化氫通過出酸管排出,液相回到罐體上口。反應溫度100-105℃,反應物料利用位差轉至後續三隻封端反應器進行保溫,第二級封端反應器反應溫度120-130℃,第三、四級封端反應器反應溫度140-150℃,各反應器物料停留時間2小時。保溫結束再經過脫酸釜熟化反應,脫酸釜溫度控制在140-150℃,脫酸釜真空度控制-0.05~0.06MPa。再經過薄膜蒸餾脫出過量的苯酚,蒸餾條件溫度150-160℃,真空度100-150pa,回收的苯酚用於套用,製得BDP粗品,其作為下一道洗滌工序的原料。
4、連續粗品精製工序
4.1連續酸洗工序
向酸洗罐內通入粗品4500kg/h及10%鹽酸800kg/h,開啟循環泵強制循環,等物料上升至酸洗罐體積的一半時,開啟酸洗上料泵,同時通入混合溶劑8000kg/h進行萃取,混合後的液體進入酸洗分層塔內分層,上層液進入分水器進一步分層,酸洗料層作為下一道工序的原料,分水器下層水排入廢水收集罐;下層水液排入廢水收集罐內,去三廢處理站處理,酸洗過程中溫度控制在60-70℃;
4.2連續鹼洗工序
酸洗結束,將有機層12500kg/h轉入中間罐中,向鹼洗罐內按配比通入酸洗好的粗品及配好的5%鹼液1800 kg/h,開啟循環泵強制循環,等物料上升至鹼洗罐體積的一半時,開啟鹼洗上料泵,混合後的液體進入鹼洗分層塔內分層,上層液進入分水器進一步分層,鹼洗料層作為下一道工序的原料,下層去廢水罐;下層水液排入廢水收集罐內,去三廢處理站處理,鹼洗過程中溫度控制在60-70℃;
4.3連續水洗工序
鹼洗結束,向水洗罐內按配比通入粗品12500kg/h及工藝水1500kg/h,開啟循環泵強制循環,等物料上升至水洗罐體積的一半時,開啟水洗上料泵,混合後的液體進入水洗分層塔內分層,上層液進入分水器進一步分層,水洗料層進入水洗粗品中間罐中,作為下一道工序的原料,分水器下層去廢水罐;下層水液排入廢水收集罐內,水洗廢水配鹼洗液用,水洗過程中溫度控制在60-70℃;
5、連續溶劑回收及過濾工序
水洗粗品經一級預熱、二級預熱後,加熱至90-100℃,通過兩級降膜蒸餾脫大部分混合溶劑,再經兩級薄膜:最後一級薄膜蒸餾在140-150℃,真空40-70pa,脫除殘留的溶劑,使產品殘留溶劑指標合格,脫出溶劑套用於酸洗,蒸出的合格成品冷卻至100-110℃,再經過濾得成品4298.3kg/h,產品收率97.52%,產品主要指標:色澤40,TPP含量0.73%,主含量N183%(HPLC)。
實施例2
與實施例1基本相同,但去掉封端反應的補加氯化鎂10Kg/h,各反應器物料停留時間3小時。其他不變,最後得成品4266.5kg/h,產品收率96.80%。產品色澤35,TPP含量0.86%,主含量N184.7%(HPLC)。
實施例3
與實施例1基本相同,但把三氯氧磷量改為5500kg/h,連續封端反應工序中的苯酚量改為2380kg,補加的氯化鎂10Kg去掉。其他不變,最後得成品4288.49kg/h,產品收率97.3%。產品色澤35,TPP含量0.78%,主含量N187.4%(HPLC)。
實施例4
與實施例1基本相同,但三氯氧磷回收採用兩級降膜蒸餾、兩級薄膜蒸餾,最後一級薄膜蒸餾條件控制真空450-500pa,物料溫度128-130℃,最後TPP含量1.02%。
實施例5
與實施例1基本相同,但苯酚回收採用一級薄膜蒸餾,控制真空280-300pa,物料溫度155-160℃,最後BDP粗品含量2500ppm;
實施例6
與實施例1基本相同,但溶劑回收採用兩級降膜蒸餾,兩級薄膜蒸餾,最後一級蒸餾條件控制真空400-500pa,物料溫度150-152℃,成品中甲苯殘留178ppm。
比較實施例(間歇法)
1、交聯反應及三氯氧磷回收工序
在搪玻璃反應釜中依次加入三氯氧磷4500kg,氯化鎂15kg。攪拌,緩慢升溫。升溫至94℃~97℃開始投雙酚A ,投料時間4小時,均勻投入。投料完畢後,升溫至115-120℃保溫,保溫時間6小時。保溫完畢,轉釜脫氧磷。開真空泵,蒸餾三氯氧磷,先低溫脫走大部分三氯氧磷,再調真空至最大繼續升溫至130~140℃,保溫至無氧磷出,蒸餾時間共約為5-7小時。
2、封端反應工序
蒸完三氯氧磷後,轉至苯酚封端反應釜,加氯化鎂10kg,在110-120℃下滴加苯酚,滴加時間大約4小時,滴加完成後,升溫至140-150℃,保溫4小時,取樣分析。合格後降溫至100℃,轉酸洗釜。
3、粗品精製工序
3.1一次酸洗:
粗品完全轉入酸洗釜後,加溶劑,升溫至65-70℃,再緩慢加入定量水1000kg,加30%鹽酸1000kg,升溫至65℃~70℃,攪拌1小時,靜置半小時分出水層;準備轉至鹼洗釜;
3.2一次鹼洗:
鹼洗釜加入水1500kg,升溫至65℃~70℃,轉入酸洗釜料,再加鹼400kg,升溫至65℃~70℃,攪拌1小時,靜置半小時分出水層;
3.3二次鹼洗:
緩慢加入水1500kg,升溫至65℃~70℃,加鹼100Kg,攪拌1小時,靜置半小時分出水層。轉至水洗釜;
3.4水洗兩次:
各緩慢加入水1500kg升溫至65℃~70℃下保溫1小時,靜置半小時分出水層,水洗二次測試PH6~7。
洗滌結束,送料至蒸餾釜脫溶劑,至溶劑大部分蒸出後,蒸餾溫度最後升至140℃~150℃,脫完後,取樣檢測溶劑含量。合格後冷卻至100℃.壓濾,得BDP成品4240.49kg,產品收率96.1%。產品色澤60,TPP含量1.13%,主含量N181.8%(HPLC)。
本發明實施例1-5與對比實施例製備BDP的能耗一覽表
從表1可以得到,本發明通過連續法製備BDP,其收率提升1-1.5%、色號提升至50以下、TPP控制在1%,比原有間歇裝置降低50%,其成本等都低於間歇法,符合高端客戶需求,且各產品質量指標波動小。