製備作為可再充電電池陽極材料的MSnx納米顆粒的方法與流程
2023-10-26 18:56:51 1
本發明總體上涉及作為可再充電電池,尤其是鈉離子或鋰離子電池的陽極材料的MSnx納米顆粒的製備方法,和包含此種材料的陽極的製備方法。
發明背景
儘管在最近幾十年中對可再充電鋰離子電池的材料已有廣泛研究,但是石墨仍然是商業電池的最廣泛使用的陽極材料。然而,石墨與許多合金型(例如Si、Ge、Sn)和轉化型材料(例如Fe3O4、MoS2、SnSb)相比具有較低比容量和體積容量(372mAhg-1;820mAhcm-3)。雖然這些材料通常經歷在鋰化/脫鋰化期間發生的大的體積變化,但是對於許多體系已經證實這種問題可以通過使用納米結構化材料緩解。[1]雖然如此,此種大容量合金型或轉化型陽極的工業化由於若干原因被阻礙。特別是對於轉化型陽極,在超過1.0V vs.Li+/Li的電勢下獲得主要份額的容量,這導致相應全電池(full-cells)的能量密度低。其次,通常電池材料的合成太成本密集或太複雜而不能以工業規模執行。在作為能夠替代商業電池中的石墨的現實備選的幾種材料中包括Sn,原因在於它兼具大多數關鍵性能:高的體積容量和比容量(~7300mAhcm-3,992mAhg-1)、低的脫鋰電勢、高的導電性和合理的價格。事實上,當前在Sony的NexelionTM電池中使用基於無定形Sn-Co-C納米複合材料的陽極,該電池已經引發對用於鋰離子電池的Co-Sn基陽極的密集研究。[2]
因此,對於日益重要的應用例如可攜式電子儀器或電動車,急迫需要替代石墨作為陽極的合適材料以改進可再充電電池尤其是鋰離子電池的能量密度。
因此需要開發允許製備MSnx納米顆粒的便宜且簡單的程序,所述MSnx納米顆粒作為可再充電電池,尤其是鋰離子電池的陽極材料顯示高的電化學性能。
技術實現要素:
因此,本發明的總體目的是提供製備MSnx納米顆粒的方法,其中
M是選自Co、Mn、Fe、Ni、Cu、In、Al、Ge、Pb、Bi、Ga的元素,和
0<x≤10,優選0<x≤3
根據本發明,製備MSnx納米顆粒的方法包括以下步驟:
-合成Sn納米顆粒:用氫化物在無水極性溶劑中的溶液還原錫鹽,將由所述溶液形成的固體Sn納米顆粒分離和洗滌所述Sn納米顆粒,
-合成M納米顆粒:用氫化物在無水極性溶劑中的溶液還原金屬鹽,將由所述溶液形成的固體M納米顆粒分離和洗滌所述M納米顆粒,
-將所述Sn納米顆粒和所述M納米顆粒機械混合以將它們轉化成MSnx納米顆粒的納米合金。
本發明的另一個目的是提供可再充電電池,尤其是鈉離子或鋰離子電池的陽極的製備方法,所述陽極包含通過本發明的方法獲得的錫基材料。
優選地,所述機械混合步驟的M納米顆粒和Sn納米顆粒的摩爾比(M/Sn)可以在1:1-1:3範圍內。
有利地,機械混合可以通過球磨達到,該球磨可以在空氣或惰性氣體中,例如在氮氣下進行。優選地,球磨在空氣中進行。
在一些優選的實施方案中,M是Co,x優選可以是大約2。
錫鹽的還原反應優選在提高的反應溫度下例如在50℃-70℃範圍內的溫度下進行。
金屬鹽的還原反應優選在更加提高的反應溫度下例如在60℃-180℃範圍內的溫度下進行,這取決於金屬鹽的反應性。
適合的氫化物的實例是NaBH4、氫化鋰、氫化鈉、氫化鉀、氫化鎂、氫化鈣、氫化三丁基錫、氫化二異丁基鋁、氫化鋰鋁、三乙基硼氫化鋰和它們的混合物。優選的氫化物是NaBH4。
無水極性溶劑的實例是1-甲基-2-吡咯烷酮(NMP),六甲基磷醯胺,1,3-二甲基-2-咪唑啉酮,1,3-二甲基-3,4,5,6-四氫-2(1H)-嘧啶酮,直鏈醚例如甘醇二甲醚、二甘醇二甲醚、三乙二醇二甲基醚,但不限於這些,亞碸例如二甲亞碸或環丁碸,但不限於這些,和它們的混合物。優選的無水極性溶劑是NMP。
適合的錫鹽的實例是氯化錫、氟化錫、溴化錫、碘化錫、錫氧化物、錫硫化物、錫酸鈉三水合物、四丁基錫和它們的混合物,優選氯化錫。
適合的合金化金屬鹽的實例是M氯化物(MCl2),M氟化物,M溴化物,M碘化物,M氧化物,M硫化物,M硫酸鹽和它們的混合物,優選Co鹽的混合物,最優選氯化Co(CoCl2)。
錫鹽或金屬鹽的還原反應各自可以在惰性氣體,優選在氮氣下進行或也可以在空氣中進行。
在優選的方法中,合成Sn納米顆粒的步驟可以包括以下步驟:
-製備氫化物在無水溶劑中的溶液和錫鹽在無水溶劑中的至少一種溶液,
-加熱所述氫化物的溶液至所述錫鹽的還原反應的反應溫度,和
-當通過將所述一種或多種錫鹽溶液快速注射到氫化物溶液中產生反應混合物而達到所述錫鹽的還原反應的反應溫度時,開始還原反應。快速意指添加以最高可能的速度並且無中斷地進行。在反應混合物中,Sn納米顆粒通過將一種或多種錫鹽添加至氫化物中而生成,並且瞬時形成。
在另一個實施方案中,合成Sn納米顆粒的步驟可以包括以下步驟:
-製備一種或多種錫鹽在無水溶劑中的溶液和氫化物在無水溶劑中的至少一種溶液,
-加熱所述錫鹽的溶液至所述錫鹽的還原反應的反應溫度,和
-當通過將氫化物溶液快速注射到所述一種或多種錫鹽的溶液中產生反應混合物而達到所述錫鹽的還原反應的反應溫度時,開始還原反應,其中Sn納米顆粒瞬時形成。
在優選的方法中,合成M納米顆粒的步驟可以包括以下步驟:
-製備氫化物在無水溶劑中的溶液和金屬鹽在無水溶劑中的至少一種溶液,
-加熱所述氫化物的溶液至所述金屬鹽的還原反應的反應溫度,和
-當通過將所述一種或多種金屬鹽的溶液快速注射到氫化物溶液中產生反應混合物而達到所述金屬鹽的還原反應的反應溫度時,開始還原反應。快速意指添加以最高可能的速度並且無中斷地進行。在反應混合物中,M納米顆粒通過將一種或多種金屬鹽添加至氫化物中生成,並且瞬時形成。
在另一個實施方案中,合成M納米顆粒的步驟可以包括以下步驟:
-製備一種或多種金屬鹽在無水溶劑中的溶液和氫化物在無水溶劑中的至少一種溶液,
-加熱所述金屬鹽的溶液至所述金屬鹽的還原反應的反應溫度,和
-當通過將氫化物溶液快速注射到所述一種或多種金屬鹽的溶液中產生反應混合物而達到所述金屬鹽的還原反應的反應溫度時,開始還原反應,其中M納米顆粒瞬時形成。
有利地,在注射後立即將已經在上述合成之一期間產生的反應混合物冷卻至室溫。更特別地,在一個優選的實施方案中,在相應地注射一種或多種錫鹽溶液或一種或多種金屬鹽的溶液後立即將通過使相應的錫鹽溶液或金屬鹽溶液與氫化物溶液合併形成的反應混合物冷卻至室溫,例如通過使用水冰浴進行冷卻。
優選地,通過離心分離將形成的固體Sn納米顆粒或M納米顆粒從它們相應的反應混合物中分離。
然後相應地洗滌所獲得的固體Sn納米顆粒或M納米顆粒,優選首先用溶劑如二甲亞碸(DMSO)然後用水洗滌。
在機械混合之前,可以在真空烘箱中在室溫乾燥Sn或M納米顆粒。
本發明方法使用簡單的製備程序,基於用於合成M-Sn基納米顆粒的便宜前體,組合了溼化學合成和機械球磨。
可以通過將上述獲得的MSnx納米顆粒、炭黑、羧甲基纖維素(CMC)和水混合來製備陽極。然後將所獲得的水性漿料塗覆在集電體上,隨後在電池組裝之前乾燥。
使用此種陽極,可以根據本領域中熟知的程序製備鋰離子電池或鈉離子電池。
附圖說明
當考慮本發明以下詳細描述時,本發明將能更好地理解並且除上面提出的那些目的以外的目的也將變得清晰。該說明將參考附圖,它們顯示:
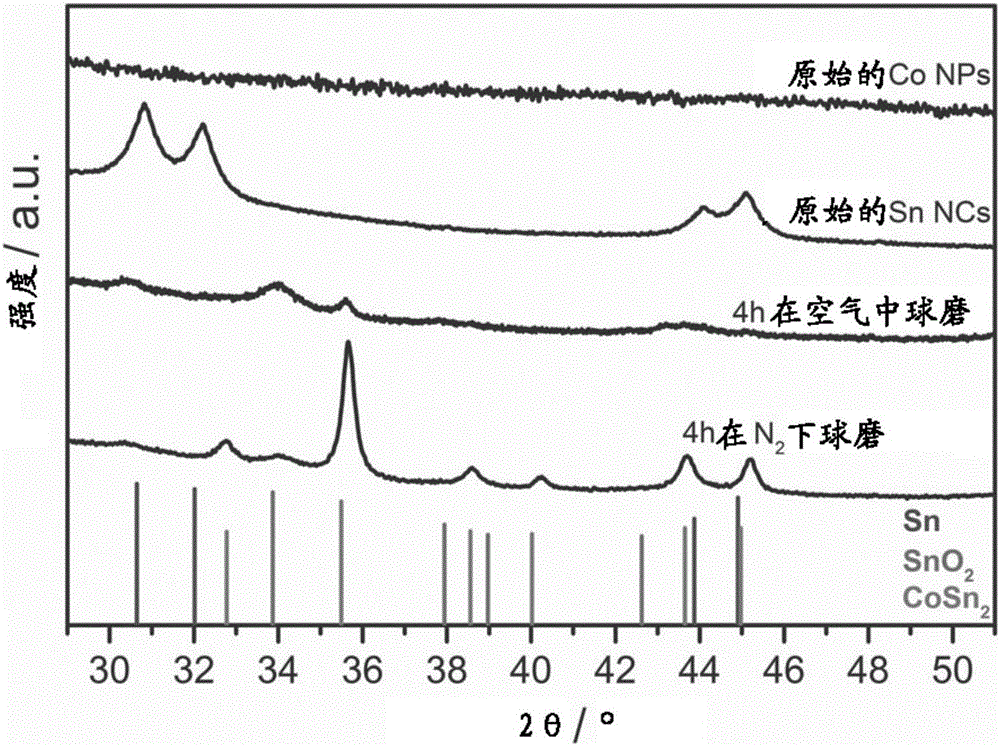
圖1.通過在空氣中球磨製備的Co NPs、Sn NCs、CoSnx NPs和通過本發明方法通過在氮氣氣氛下球磨製備的CoSn2 NCs的X射線衍射(XRD)圖案。
圖2.無定形Co NPs的EDX-譜。
圖3.Co NPs(A)、Sn NCs(B)、CoSnx NPs(C)和CoSn2 NCs(D)的透射電子顯微(TEM)圖像。
圖4.在鋰離子半電池中在1984mAg-1的電流下在0.005-1.0V的電勢範圍內Sn NCs、CoSn2 NCs和CoSnx NPs的容量保持。
圖5.對應於圖4的Sn NCs(A)、CoSn2 NCs(B)和CoSnx NPs(C)的電流充電/放電曲線。
圖6.在鋰離子半電池中使用0.1Vs-1的掃描速率在0.005-1.0V電勢範圍中試驗的Sn NCs(A)、CoSn2 NCs(B)和CoSnx NPs(C)的循環伏安圖。
圖7.在鋰離子半電池中在0.005-1.0V電勢範圍內Sn NCs、CoSn2 NCs和CoSnx NPs的倍率性能(rate capability)測量。
圖8.(A)用LiCoO2作為陰極材料的CoSnx NPs鋰離子全電池在500mAg-1電流下的容量保持,和(B)與(A)對應的CoSnx/LiCoO2全電池的電流充電/放電曲線,以平均放電電壓作為插入圖(inset)。
具體實施方式
根據本發明方法,首先通過用氫化物在無水極性溶劑中的溶液還原相應的金屬氯化物分開合成Sn納米顆粒(NPs)和M NPs。
在Sn或M NPs,尤其是Co NPs的典型的合成中,將適合量的氫化物例如NaBH4溶解在合適量的無水極性溶劑例如1-甲基-2-吡咯烷酮(NMP)中,在攪拌的同時加熱。為了合成Sn NPs,在達到所需的溫度,例如60℃後,迅速地注射錫鹽,例如SnCl2在無水溶劑例如NMP中的溶液。對於合成M NPs,在達到所需的溫度,例如對於M=Co而言150℃或對於M=Mn、Fe、Ni而言120℃後,迅速地注射金屬鹽,例如CoCl2、MnCl2、FeCl2、NiCl2在無水溶劑例如NMP中的溶液。
立即形成固體Sn NPs或M NPs。在注射後,將各自的懸浮液冷卻至室溫,例如用水冰浴冷卻。通過離心分離使相應獲得的材料從它們的溶液中分離並用二甲亞碸(DMSO)洗滌一次和用水洗滌兩次以除去未反應的NaBH4和水溶性副產物例如NaCl。最後可以在真空烘箱中在室溫下乾燥相應的反應產物。通常,反應按如下量得到所需產物:對於Sn NPs 67%,對於Co NPs 98%,對於Fe NPs 36%,對於Mn NPs 16%和對於Ni NPs 34%。
在上述方法中,以下化學物質適當地通用於獲得Sn NPs或M NPs。然而,為了考慮MCl2和SnCl2的不同反應性,使用不同的反應溫度和前體濃度:
I.作為除NMP以外的無水溶劑:
任何醯胺例如六甲基磷醯胺、1,3-二甲基-2-咪唑啉酮或1,3-二甲基-3,4,5,6-四氫-2(1H)-嘧啶酮。
II.作為NaBH4的替代:
任何鹼金屬或鹼土金屬氫化物例如氫化鋰、氫化鈉、氫化鉀、氫化鎂、氫化鈣或其它金屬氫化物例如氫化三丁基錫、氫化二異丁基鋁、氫化鋰鋁或三乙基硼氫化鋰。
III.作為SnCl2的替代:
任何錫滷化物例如氟化錫、溴化錫、碘化錫;任何錫氧化物、錫硫化物、錫酸鈉三水合物或四丁基錫。
IV.作為MCl2的替代:
任何金屬滷化物例如金屬氟化物、金屬溴化物、金屬碘化物;任何金屬氧化物、金屬硫化物或金屬硫酸鹽。
上述化學物質可以單獨使用或與它們相應I-IV組中的一種或多種其它成員組合使用。
顆粒尺寸可能受數種特性例如氫化物量、反應溫度和冷卻速度的影響。
取決於採用的NaBH4的量,NPs的尺寸可以改變,即NaBH4的量越高,顆粒越小。另外,為了以高產率製備小尺寸的NPs,需要過量的NaBH4。
Sn NPs或M NPs也可以在室溫下製備,然而,較小NPs的形成很可能在高溫下是有利的。
可以如下獲得具有大致5-10nm的直徑的Sn NPs:採用大約96倍過量的NaBH4作為還原劑並在SnCl2的注射後直接地急速冷卻。
可以如下獲得具有大致4-10nm的直徑的M NPs:採用大約4-8倍過量的NaBH4作為還原劑並在MCl2的注射後直接地急速冷卻。
可以通過本領域普通技術人員已知的任何適當的冷卻技術實現急速冷卻。
另外,已經發現,Sn或M NPs的此種合成可以在空氣中進行,這顯著地降低成本(材料以及工作時間)。
除容易進行和相當便宜之外,製備本發明中使用的Sn NPs和M NPs的方法與文獻中描述的方法相比具有若干優點。這種合成程序的優點以下:
I.不需要使用表面活性劑。
II.反應也可以在空氣中進行。
III.不貴且安全的化學物質:這裡優選使用的NaBH4是最便宜的金屬氫化物,
IV.洗滌程序:所獲得的Sn或M NPs簡單地用水在空氣中洗滌。
V.取決於所使用的NaBH4的過量和反應溫度,可以調整顆粒的尺寸。
在合成Sn NPs和M NPs後,製備Sn NPs和M NPs的混合物。這些混合物中的M納米顆粒和Sn納米顆粒的摩爾比在1:1至1:3範圍內。
在空氣中或在氮氣下球磨Sn NPs和M NPs的混合物,目標是將材料合金化,獲得MSnx納米顆粒。有利地,以1800-2400rpm的頻率將M納米顆粒和Sn納米顆粒球磨2-4小時。
可以通過將MSnx NPs、炭黑、CMC和水混合,優選通過使用球磨機混合例如1h製備陽極。然後將所獲得的水性漿料塗覆在集電體如Cu集電體上,隨後在電池組裝之前乾燥,例如在80℃下在真空下乾燥過夜。
實驗部分
I.所使用的材料
化學物質和溶劑:氯化錫SnCl2(99.9%,Alfa Aesar)、CoCl2(98%,Sigma-Aldrich)、1-甲基-2-吡咯烷酮(NMP,無水,99.5%,Fisher BioReagents)。
電池組分:炭黑(CB,Super C65,由TIMCAL提供),羧甲基纖維素(CMC,品級:2200,Daicel Fine Chem Ltd.);氟代碳酸亞乙酯(FEC,Solvay,電池級),LiPF6在碳酸亞乙酯/碳酸二甲基酯中的1M溶液(EC:DMC:1:1,Merck,電池級),玻璃微纖維隔片(GF/D,Whatman,Cu箔(9μm,MTI Corporation)
II.方法
CoSnx NPs的合成
實施例1:CoSn2納米晶體(NCs)的合成
根據本發明,CoSn2 NCs的合成包括合成Sn NCs,合成Co NPs,和通過球磨Sn NCs和Co NPs合成CoSn2 NCs:
合成Sn NCs:
在Sn NCs的典型合成中,在氮氣下將96mmol NaBH4溶解在85mL無水NMP中並加熱到60℃,同時機械攪拌。在達到60℃後,迅速地注射1mmol SnCl2預先溶解在無水NMP中的溶液並使用水冰浴立即將該反應混合物冷卻至室溫。通過離心分離從溶液分離反應產物,用二甲亞碸洗滌一次,然後用水洗滌兩次。獲得Sn NCs。
反應產量是80mg(67%)。所獲得的產物的XRD圖案僅顯示對應於結晶β-Sn的峰(圖1,索引到四方晶系Sn,空間群I41/amd(141),ICDD PDF條目號:00-004-0673)。反應產物的TEM分析顯示顆粒是多分散的,具有5-10nm的尺寸(圖3B)。
合成Co NPs:
在Co NPs的典型合成中,使用與Sn NCs相似的程序,但有改變,尤其是反應溫度和前體濃度):在氮氣下將32mmol NaBH4溶解在15mL無水NMP中並加熱到150℃,同時機械攪拌。在達到150℃後,迅速地注射8mmol CoCl2預先溶解在無水NMP中的溶液並使用水冰浴立即將該反應混合物冷卻至室溫。通過離心分離從溶液分離反應產物,用二甲亞碸洗滌一次,然後用水洗滌兩次。獲得Co NPs。
反應產量是460mg(98%)。所獲得的產物的XRD圖案顯示獲得無定形的Co NPs。圖2示出了無定形Co NPs的EDX-譜。對應於S(樣品的大約1wt%)的峰可能歸因於殘留DMSO。反應產物的TEM分析顯示顆粒是多分散的,具有4-7nm範圍的尺寸(圖3A)。
合成CoSn2納米晶體(NCs):
為了製備Co-Sn基NCs,以30s-1的頻率將如上製備的1.4mmol Sn NCs與如上製備的0.7mmol Co NPs球磨4小時。在氮氣氣氛下加載用於球磨的燒杯並密封。
反應產量是200mg(96%)。所獲得的產物的XRD圖案顯示在惰性條件下球磨導致形成結晶的CoSn2納米合金(參考圖案:四方晶系SnO2,空間群P42/mnm(136),ICDD PDF條目00-077-0448;四方晶系CoSn2,空間群I4/mcm(140),ICDD PDF條目00-025-0256)。反應產物的TEM分析顯示顆粒是多分散的,具有6-20nm的尺寸(圖3D)。
實施例2:合成CoSnx納米顆粒(NPs)
為了製備Co-Sn基NCs,以30s-1的頻率將如上製備的1.4mmol Sn NCs與如上製備的0.7mmol Co NPs球磨4小時。在空氣中加載用於球磨的燒杯。
反應產量是200mg(96%)。所獲得的產物的XRD圖案顯示在空氣中球磨導致形成無定形的CoSnx NPs。樣品的主要級分無定形化,僅具有在34°對應於SnO2和在35.5°對應於CoSn2的小特徵。反應產物的TEM分析顯示顆粒是多分散的,具有6-20nm的尺寸(圖3C)。
III.製備Co-Sn基電極、電池組裝和電化學測量
為了電極製備,通過以500rpm球磨1小時將相應的NPs(64wt.%)與CB(21wt%)、CMC(15wt%)和作為溶劑的水混合來製備水性漿料。將所得的漿料塗覆到銅集電體上,在電池組裝之前在真空下在80℃乾燥12小時。在於Ar填充的手套箱(O2<0.1ppm,H2O<0.1ppm)中組裝的氣密性硬幣型電池中進行電化學測量,對於鋰離子半電池試驗使用元素鋰或對於鋰離子全電池試驗使用在鋁箔(MTI)上的LiCoO2。使用一片玻璃微纖維作為隔片。作為電解質,使用在具有3%FEC的EC:DMC中的1M LiPF6。將FEC添加到電解質中以改進循環穩定性。電流循環試驗在室溫下在MPG2多路工作站(BioLogic)上進行。通過用於半電池和全電池試驗的Co-Sn納米顆粒的質量(不包括CB和粘結劑)將容量標準化。
IV.表徵
使用碳塗覆的Cu柵格作為基底(Ted-Pella)用Philips CM30 TEM顯微鏡在300kV下獲得透射電子顯微(TEM)圖像。使用NanoSEM 230進行能量分散X射線光譜(EDX)測量。在STOE STADI P粉末X射線衍射儀上測量粉末X射線衍射(XRD)。
V.電化學結果
圖4示出了Co-Sn基NPs在0.005–1.0V電勢範圍中在1984mAg-1的高電流下經1500個循環的容量保持。1984mAg-1的電流對於Sn而言相當於2C倍率,基於形成Li4.4Sn的992mAhg-1的理論容量。假定Co不貢獻於容量,則CoSnx NPs和CoSn2 NCs的理論容量為795mAhg-1,因此與純Sn相比更低。對於恆電流循環試驗,上限截止(cut-off)電勢限於1.0V,從而僅包括與全電池中的高能量密度對應的過程。如可從圖4中在1984mAg-1下的恆電流循環看出的那樣,在首次100個循環期間在容量快速增加後,Sn NCs以及CoSnx NPs和CoSn2 NCs顯示~570mAhg-1的容量。然而Sn NCs在400個循環後顯示明顯的容量衰減,Co-Sn基NPs顯示好得多的容量保持。特別地,對於CoSn2 NCs,在1500個循環後,仍然保持462mAhg-1。對於CoSnxNPs,容量保持甚至更好,為525mAhg-1,這相當於在1984mAg-1的電流下經1500個循環僅8%的衰減。因此,通過本發明方法獲得的CoSnx NPs具有超高的循環穩定性。包括Co的CoSnxNPs與純Sn NCs相比的優異循環穩定性可能歸因於兩個作用。由於Co不形成鋰合金這一事實,它可以在循環期間充當非活性基體並因此緩衝由Sn的鋰化/脫鋰化引起的體積變化。此外,Co的存在可以防止Sn NCs聚集並因此進一步改進循環穩定性。CoSn2 NCs和CoSnx NPs之間在循環穩定性方面的差異可能歸因於這樣的事實:CoSnx NPs由於它們通過球磨在空氣中製備而更加氧化。更高含量的氧化物可能導致形成更加有效的Li2O基體以緩衝體積變化和抑制Sn域在循環期間的燒結。應該指出的是,對於所有三個體系,在第一放電循環由於固體電解質形成而獲得~30%的值之後,平均庫侖效率在循環期間是99.6%。Sn NCs、CoSn2NCs和CoSnx NPs的平均脫鋰電勢同樣低,在循環期間具有~0.5V vs.Li+/Li的穩定值(圖5A至5C和圖6A至6C)。
為了評價Co-Sn基NPs的倍率性能,進行在0.2C-10C之間的電流倍率下的恆電流循環試驗(圖7,1C=992mAg-1)。由於在所有情況下NPs的小尺寸和因此增強的反應動力學,觀察到類似的好的倍率性能,不過對於0.5C–2C的電流,Sn NCs顯示與CoSn2 NCs相比容量低~50mAhg-1。對於高達10C的倍率,所有三種材料仍然保持~350mAhg-1的容量。引起興趣地,可以觀察到,在如此高電流下,容量在循環期間增加,導致在所述倍率逐步降回到0.2C期間獲得相同或甚至更高的容量。特別是對於CoSnx NPs,初始在0.5C–2C的倍率下觀察到的在容量方面與CoSn2 NCs的微小差異在循環期間完全減小。
為了試驗Co-Sn基NPs在更加實際的條件下的應用性,進行使用LiCoO2作為陰極的陽極限制的全電池試驗。選擇CoSnx NPs作為陽極材料,原因在於它們與Sn和CoSn2 NCs相比優異的容量保持。這裡,所有容量和電流與CoSnx NPs的質量有關。最初將CoSnx NPs/LiCoO2的全電池充電到2000mAhg-1以考慮在第一循環中的不可逆充電損失。對於隨後的循環,充電和放電限於500mAhg-1。在這些條件下以500mAg-1的電流循環,CoSnx NPs顯示穩定的容量,對於50個循環具有3.2V的平均放電電壓。基於陽極容量和放電電壓,大致估計CoSnx NPs的比能量密度與石墨相當(372mAhg-1,3.6V vs.LiCoO2)。然而,考慮到本體β-Sn(~7.3gcm-3)和Co(~8.9gcm-3)與石墨(~2.2gcm-3)相比高得多的密度,使用CoSnx NPs可潛在地提高體積能量密度高達4倍。
圖8示出了用LiCoO2作為陰極材料的CoSnx NPs鋰離子全電池的電化學性能:圖8A:在500mAg-1電流下的容量保持。圖8B:與(A)對應的SnSb/LiCoO2全電池的恆電流充電/放電曲線,以平均放電電壓作為插入圖(inset)。限制充電和放電容量到500mAhg-1的情況下循環電池。顯示的容量和電流與CoSnx NPs的質量有關。
總之,本發明的方法允許通過用在NMP中的NaBH4簡單還原相應的金屬氯化物合成具有直徑≤10nm的Co NPs和Sn NCs,隨後通過球磨將它們轉化成金屬間結晶和無定形的Co-Sn納米合金。儘管CoSn2 NCs顯示對於數百循環的好的循環穩定性,但是無定形的CoSnxNPs顯示優異的容量保持,在1984mAg-1下經1500個循環僅有8%衰減。另外,用LiCoO2作為陰極材料在鋰離子全電池中試驗,CoSnx NPs提供500mAhg-1的穩定容量,平均放電電壓為3.2V。考慮到便宜和容易大規模化的製備方法和它們的由高可循環性以及高體積和比能量密度表徵的優異電化學性能,這裡提供的CoSnx NPs作為Li離子和Na離子電池的高性能陽極材料具有巨大潛能。
雖然已顯示和描述了本發明目前的優選實施方案,但是應清楚地理解,本發明不限於它們,而是可以在權利要求書範圍內以其它方式多樣地實施和實踐。
參考文獻
[1]a)P.G.Bruce,B.Scrosati,J.-M.Tarascon,Angew.Chem.Int.Ed.2008,47,2930-2946;b)M.F.Oszajca,M.I.Bodnarchuk,M.V.Kovalenko,Chem.Mater.2014,26,5422-5432。
[2]a)A.D.W.Todd,R.E.Mar,J.R.Dahn,J.Electrochem.Soc.2007,154,A597-A604;b)X.-L.Wang,W.-Q.Han,J.Chen,J.Graetz,ACS Appl.Mater.Interfaces 2010,2,1548-1551。