芳基烷基硫醚類衍生物及其製備方法與流程
2023-10-31 02:24:57 4
本發明涉及有機合成領域,且特別涉及一種芳基烷基硫醚類衍生物及其製備方法。
背景技術:
芳基烷基硫醚類衍生物廣泛應用於醫藥、染料、輕化用品、農業、高聚材料等領域。一些具有生物活性的芳基烷基硫醚化合物在醫藥分子研究中展示了其獨特的藥理作用,例如含有芳基烷基硫醚結構單元的化合物i能有效抑制使免疫系統產生缺陷的病毒蛋白酶(hivpr)的生物活性,是治療愛滋病的重要藥物。
隨著有機硫化合物在合成中的廣泛應用,近年來有機硫化合物在有機合成中的應用得到普遍重視。芳基烷基硫醚類化合物的合成路徑引起了眾多有機化學家的積極思考,並且得出了一些有效的方法。通過芳環碳或者烷基碳與硫原子的c―s偶聯是芳基烷基硫醚類化合物合成的主要途徑,過渡金屬催化被廣泛應用於芳基烷基硫醚類化合物的c―s偶聯反應。
過渡金屬催化合成芳基烷基硫醚的三種主要方法中,鈀類催化劑具有較高的毒性以及不菲的價格,並且對含磷的配體具有依賴制。銅、鎳類催化劑也需在加入配體及其它添加劑的情況下才能發揮催化作用,而額外試劑的加入必然也對反應條件及原料分子結構中基團的兼容性起了一定的限制作用。因此,高效實用並具有原子經濟性的綠色合成方法在c―s偶聯反應製備芳基烷基硫醚類化合物的研究中成為了一個新的導向。
技術實現要素:
本發明的目的在於提供一種芳基烷基硫醚類衍生物,該多環芳硫醚衍生物有多環的存在,其結構更加複雜多樣,在化工生產、臨床醫藥中也將表現出更加廣闊的用途前景。
本發明的另一目的在於提供一種芳基烷基硫醚類衍生物的製備方法,該方法克服了以往反應中路線過長,底物和反應條件要求苛刻,取代官能團擴展有限的等缺點。
本發明解決其技術問題是採用以下技術方案來實現的:
本發明提供一種芳基烷基硫醚類衍生物,其結構式為:
其中,e=co2r,r選自直鏈烷基、支鏈烷基、烯基、炔基或者芳香烴基團中任意一種基團;r1選自氫基、滷素基團、直鏈烷基、支鏈烷基、酯基或者烷氧基及其衍生物中的任意一種基團,r1可處於苯環的任意位置;r2選自直鏈烷基、支鏈烷基或者芳香烴基團及其衍生物中任意一種基團。
本發明還提供一種芳基烷基硫醚類衍生物的製備方法,包括以下步驟:在無水無氧體系中,將二炔類化合物、烴基炔溴、無水乙腈、過渡金屬催化劑和有機鹼混合併反應得到中間體。在90-100℃條件下,將中間體、烷基二硫醚和甲苯混合併反應。
本發明芳基烷基硫醚類衍生物及其製備方法的有益效果是:本發明提供的一系列新的芳基烷基硫醚類衍生物相對於普通硫醚衍生物有多環的存在,其結構更加複雜多樣,在化工生產、臨床醫藥中也將表現出更加廣闊的用途前景。本發明的製備方法克服了以往反應中路線過長,底物和反應條件要求苛刻,取代官能團擴展有限的等缺點,該反應不但底物合成簡單、試劑比較便宜,並且具有高原子經濟性、綠色環保的得到目標分子,給芳基烷基硫醚類衍生物的工業化生產提供了一條很有價值的途徑。
附圖說明
為了更清楚地說明本發明實施例或現有技術中的技術方案,以下將對實施例中所需要使用的附圖作簡單地介紹。
圖1為本發明實施例1製備得到的芳基烷基硫醚類衍生物的核磁共振氫譜;
圖2為本發明實施例1製備得到的芳基烷基硫醚類衍生物的核磁共振碳譜;
圖3為本發明實施例2製備得到的芳基烷基硫醚類衍生物的核磁共振氫譜;
圖4為本發明實施例2製備得到的芳基烷基硫醚類衍生物的核磁共振碳譜;
圖5為本發明實施例3製備得到的芳基烷基硫醚類衍生物的核磁共振氫譜;
圖6為本發明實施例3製備得到的芳基烷基硫醚類衍生物的核磁共振碳譜;
圖7為本發明實施例4製備得到的芳基烷基硫醚類衍生物的核磁共振氫譜;
圖8為本發明實施例4製備得到的芳基烷基硫醚類衍生物的核磁共振碳譜;
圖9為本發明實施例4製備得到的芳基烷基硫醚類衍生物的單晶x-ray圖。
具體實施方式
為使本發明實施例的目的、技術方案和優點更加清楚,下面將對本發明實施例中的技術方案進行清楚、完整地描述。實施例中未註明具體條件者,按照常規條件或製造商建議的條件進行。所用試劑或儀器未註明生產廠商者,均為可以通過市售購買獲得的常規產品。
在本發明的描述中,需要說明的是,術語「第一」、「第二」等僅用於區分描述,而不能理解為指示或暗示相對重要性。
下面對本發明實施例的石墨烯-卟啉類有機物納米材料的製備方法以及石墨烯-卟啉類有機物納米材料進行具體說明。
本發明實施例提供的芳基烷基硫醚類衍生物及其製備方法進行具體說明。
本發明實施例提供的一種芳基烷基硫醚類衍生物,其結構式為:
其中,e=co2r,r選自直鏈烷基、支鏈烷基、烯基、炔基或者芳香烴基團中任意一種基團;r1選自氫基、滷素基團、直鏈烷基、支鏈烷基、酯基或者烷氧基及其衍生物中的任意一種基團,r1可處於苯環的任意位置;r2選自直鏈烷基、支鏈烷基或者芳香烴基團及其衍生物中任意一種基團。
本發明實施例中r中的直鏈烷基選自甲基、乙基、正丙基、正丁基、正戊基、正己基、正庚基、正辛基、正壬基、正癸基或者其他無支鏈的烷基中的任意一種基團。優選地r中的直鏈烷基為乙基。r中的支鏈烷基選自異丙基、異丁基、叔丁基、異戊基、叔戊基、新戊基、異己基、叔己基、新己基等具有支鏈的烷基中的任意一種基團。進一步優選地,r中的支鏈烷基為異丙基。r中的烯基可以是具有支鏈的烯基也可以是不具有支鏈的烯基。r中的炔基可以是具有支鏈的烯基也可以是不具有支鏈的炔基或者是二者的衍生物。r中的芳香烴基團可以是芳香烷烴也可為芳香烷烴的衍生物或者是二者的衍生物。
在本發明實施例中r1中的滷素基團選自基團、氯基、溴基或者碘基中任意一種基團。進一步優選地,r1中的滷素基團為氟基。r1中的直鏈烷基選自甲基、乙基、正丙基、正丁基、正戊基、正己基、正庚基、正辛基、正壬基、正癸基或者其他無支鏈的烷基中的任意一種基團。進一步優選地,r1中的直鏈烷基選自甲基或者乙基。r1中的支鏈烷基選自異丙基、異丁基、叔丁基、異戊基、叔戊基、新戊基、異己基、叔己基、新己基等具有支鏈的烷基中的任意一種基團。r1中的烷氧基可以是甲氧基、乙氧基、正丙氧基、異丙氧基、正丁氧基、異丁氧基、叔丁氧基、正戊氧基、異戊氧基、叔戊氧基、新戊氧基或者其他的烷氧基團。進一步優選地,r1中的烷氧基為甲氧基。
在本發明實施例中r2中的直鏈烷基選自甲基、乙基、正丙基、正丁基、正戊基、正己基、正庚基、正辛基、正壬基、正癸基或者其他無支鏈的烷基中的任意一種基團。r2中的支鏈烷基為異丙基、異丁基、叔丁基、異戊基、叔戊基、新戊基、異己基、叔己基、新己基等具有支鏈的烷基中的任意一種基團。r2中的芳香烴基團選自對氯苯基、間氟苯基、鄰溴苯基或苄基等含有苯環的基團中任意一種基團。進一步優選地,r2中的芳香烴基團為苄基。
本發明實施例還提供一種芳基烷基硫醚類衍生物的製備方法:
具體的反應式為:
具體的合成包括以下步驟:
s1、製備中間體;
在無水無氧體系中,將二炔類化合物和烴基炔溴加入到無水乙腈中,而後加入過渡金屬催化劑和有機鹼混合併在室溫下攪拌12-20小時反應得到中間體。
採用無水無氧體系是為了保證過渡金屬催化劑的活性,若反應體系中存在水或者氧氣將會使得過渡金屬催化劑失去活性,進而不能合成得到中間體,影響芳基烷基硫醚類衍生物的生成。而採用過渡金屬作為催化劑降低了反應所需的能量,使得反應能夠順利進行。優選地,所用的二炔類化合物為碳原子橋連的二炔類化合物。
優選地,過渡金屬催化劑為pd(pph3)2cl2和cui,pd(pph3)2cl2和cui的物質的量之比為2.8-3.2:1。採用pd(pph3)2cl2和cui混合作為催化劑時,pd(pph3)2cl2作為催化劑,而cui作為助催化劑,提升催化劑的效能,加快反應速率,提高產率。二者使用的比例保證了催化效果,進一步地加快反應速率,減少副產物。
優選地,有機鹼為胺類化合物,例如甲胺、乙胺、丙胺、丁胺、戊胺、二乙胺、二丙胺、二丁胺、二戊胺、二己胺、三乙胺等。進一步優選地有機鹼為三乙胺。三乙胺可以反應生成的溴化氫反應,進而促進整個反應平衡向生成物方向進行。
進一步優選地,二炔類化合物、烴基炔溴、過渡金屬催化劑和有機鹼的物質的量之比為1:2-3:0.03-0.05:2-5,二炔類化合物在無水乙腈中的濃度為0.5-0.8mol/l。在該優選值範圍內進行的反應速率快,產率高,副反應少。
s2、純化中間體;
純化中間體,是去除中間體內可能含有的雜質,減少雜質對後續反應的影響,同時減少芳基烷基硫醚類衍生物內可能含有的雜質。甚至中間體內含有的雜質可能導致後續不能反應生成得到芳基烷基硫醚類衍生物。
純化是將二炔類化合物、烴基炔溴、無水乙腈、過渡金屬催化劑和有機鹼混合併反應得到的第一反應液首先採用質量百分比為15%的鹽酸溶液洗滌去除有機鹼,而後再用質量百分比為10%的碳酸氫鈉溶液洗滌去除鹽酸溶液中的鹽酸,再用飽和食鹽水洗滌以去除碳酸氫鈉。接著採用乙酸乙酯萃取洗滌後第一反應液,除去水相。然後使用無水硫酸鎂乾燥萃取後得到的有機相。乾燥後使用旋轉蒸發儀旋蒸以去除有機相中的有機溶劑。最後再用柱層析純化旋蒸後得到物質,得到純度高的中間體。其中,柱層析採用的淋洗劑為乙酸乙酯與石油醚的混合液,混合液中乙酸乙酯與石油醚的體積比為1:40-60。採用上述的淋洗液能夠良好的分離中間體和雜質。
s3、合成得到芳基烷基硫醚類衍生物粗品;
在90-100℃條件下,將中間體和烷基二硫醚在甲苯溶劑中攪拌混合反應18-24小時,得到芳基烷基硫醚類衍生物的粗產物。溫度為影響該反應的重要因素,若溫度高於或低於該溫度範圍則生成物質的結構將發生變化,得不到本發明所需的芳基烷基硫醚類衍生物。
優選地,中間體與烷基二硫醚的物質的量之比為1:1-1.5。甲苯作為溶劑時,中間體在甲苯中的濃度是0.1-0.3mol/l。在該優選值範圍內進行的反應速率快,產率高、副產物少。
s4、純化芳基烷基硫醚類衍生物粗品;
反應得到的芳基烷基硫醚類衍生物粗品內含有雜質,這些雜質會影響對芳基烷基硫醚類衍生物結構的鑑定或者影響後續的使用。因此,需要對其進行進一步地純化。
將製備得到的芳基烷基硫醚類衍生物粗品即第二反應液經過去離子水洗滌後,用乙酸乙酯萃取,用無水硫酸鎂乾燥萃取液。過濾後,用旋轉蒸發儀蒸乾萃取液中溶劑。芳基烷基硫醚類衍生物粗品用柱層析的方法分體提純,柱層析的淋洗劑為乙酸乙酯與石油醚的混合液,混合液中乙酸乙酯與石油醚的體積比為1:30-60,柱層析產率約為75%。
本發明實施例製備得到的芳基烷基硫醚類衍生物的結構通過1hnmr和13cnmr來測定。
本發明實施例提供的一系列新的芳基烷基硫醚類衍生物相對於普通硫醚衍生物有多環的存在,其結構更加複雜多樣。並且,本發明實施例提供的製備方法簡便、高效、綠色環保,在化工生產,給芳基烷基硫醚類衍生物的工業化生產提供了一條很有價值的途徑。
以下結合實施例對本發明的特徵和性能作進一步的詳細描述。
實施例1
本實施例提供的一種芳基烷基硫醚類衍生物,其結構式如(i)式所示。
將丙二酸二乙酯橋連的二炔化合物(20mmol)與苯乙炔溴(48mmol)加入到反應瓶中,混合在pd(pph3)2cl2和cui作為催化劑的無水無氧催化體系中,其中催化劑的量為0.8mmol,pd(pph3)2cl2:cui=3:1。以三乙胺(50mmol)作鹼,以無水乙腈(40ml)為溶劑,室溫下攪拌反應12小時,將反應產物先後用15%的鹽酸溶液、10%的碳酸氫鈉溶液、飽和食鹽水分別洗滌;然後用乙酸乙酯萃取,減壓旋幹,柱層析(淋洗劑中乙酸乙酯與石油醚的體積比為1:40)得到淺棕色固體產物。
在95℃的條件下,將中間體(1mmol)與二丙基二硫醚(1.2mmol)加入到5毫升甲苯溶劑中,反應20小時,得到芳基烷基硫醚衍生物粗品。
將芳基烷基硫醚衍生物粗品加水洗滌後,用乙酸乙酯萃取,減壓旋幹,柱層析(淋洗劑中乙酸乙酯與石油醚的體積比為1:40)分離可得到純化的目標產物,即芳基烷基硫醚衍生物,柱層析產率約為76%。
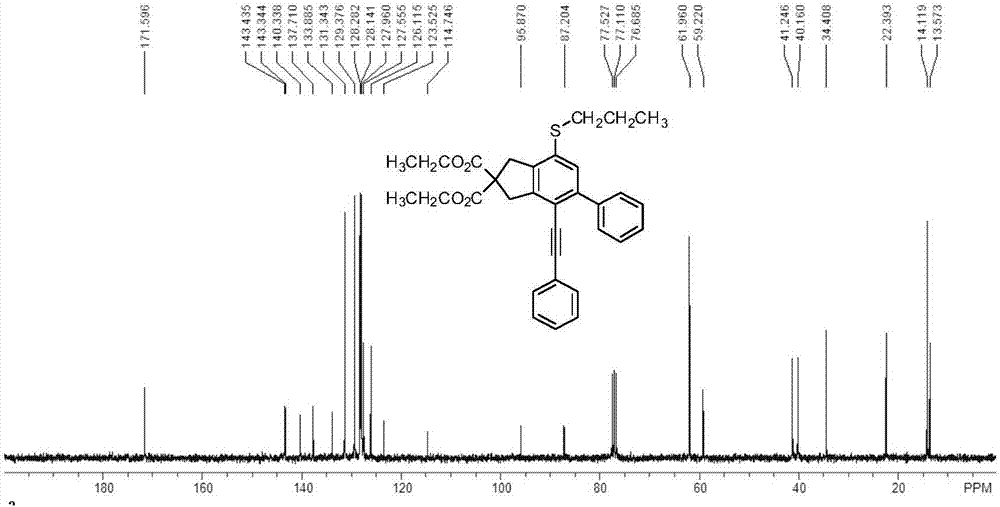
芳基烷基硫醚衍生物的結構通過1hnmr和13cnmr來測定,具體圖譜見圖1和圖2。
白色固體產物:
1hnmr(300mhz,cdcl3):δ7.63―7.61(d,2h,j=6.6hz),7.45―7.38(m,3h),7.33―7.27(m,5h),7.12(s,1h),4.28―4.21(q,4h,j=7.2hz),3.83(s,2h),3.63(s,2h),2.96―2.92(t,2h,j=7.2hz),1.75―1.64(m,2h),1.31―1.26(t,6h,j=7.2hz),1.07―1.02(t,3h,j=7.2hz);
13cnmr(75.5mhz,cdcl3):δ171.6,143.4,143.3,140.3,137.7,133.9,131.3,129.4,128.3,128.1,128.0,127.6,126.1,123.5,114.7,95.9,87.2,62.0,59.2,41.2,40.2,34.4,22.4,14.1,13.6。
實施例2
本實施例提供的一種芳基烷基硫醚類衍生物,其結構式如(ii)式所示。
本實施例還提供一種製備上述芳基烷基硫醚類衍生物的方法:
將丙二酸二異丙酯橋連的二炔化合物(20mmol)與對甲氧基苯乙炔溴(40mmol)加入到反應瓶中,混合在pd(pph3)2cl2和cui作為催化劑的無水無氧催化體系中,其中催化劑的量為0.6mmol,pd(pph3)2cl2:cui=3.2:1。以三乙胺(60mmol)作鹼,以無水乙腈(25ml)為溶劑,室溫下攪拌反應20小時,將反應產物先後用15%的鹽酸溶液、10%的碳酸氫鈉溶液、飽和食鹽水分別洗滌;然後用乙酸乙酯萃取,減壓旋幹,柱層析(淋洗劑中乙酸乙酯與石油醚的體積比為1:50)得到淺棕色固體產物。
在90℃的條件下,將中間體(1mmol)與二苄基二硫醚(1mmol)加入到3.5毫升甲苯溶劑中,反應22小時,得到芳基烷基硫醚類衍生物粗品。
將芳基烷基硫醚類衍生物粗品加水洗滌後,用乙酸乙酯萃取,減壓旋幹,柱層析(淋洗劑中乙酸乙酯與石油醚的體積比為1:30)分離可得到純化的目標產物,即芳基烷基硫醚類衍生物,柱層析產率約為71%。
芳基烷基硫醚類衍生物的結構通過1hnmr和13cnmr來測定,具體檢測圖譜見圖3和圖4。
白色固體產物:
1hnmr(300mhz,cdcl3):δ7.50―7.47(d,2h,j=8.4hz),7.31―7.25(m,7h),7.08(s,1h),6.96―6.93(d,2h,j=8.7hz),6.84―6.81(d,2h,j=8.7hz),5.12―5.03(m,2h),4.14(s,2h),3.86(s,3h),3.80(s,3h),3.77(s,2h),3.56(s,2h),1.27―1.25(d,12h,j=6.3hz);
13cnmr(75.5mhz,cdcl3):δ171.1,159.5,159.1,143.3,142.6,138.2,136.9,132.8,132.7,132.3,130.4,129.0,128.6,127.9,127.4,115.9,115.7,113.9,113.3,96.2,85.9,69.3,59.2,55.4,55.3,41.2,40.1,38.0,21.6。
實施例3
本實施例提供的一種芳基烷基硫醚類衍生物,其結構式如(iii)式所示。
將丙二酸二異丙酯橋連的二炔化合物(20mmol)與對氟苯乙炔溴(60mmol)加入到反應瓶中,混合在pd(pph3)2cl2和cui作為催化劑的無水無氧催化體系中,其中催化劑的量為1.0mmol,pd(pph3)2cl2:cui=2.8:1。以三乙胺(40mmol)作鹼,以無水乙腈(33ml)為溶劑,室溫下攪拌反應16小時,將反應產物先後用15%的鹽酸溶液、10%的碳酸氫鈉溶液、飽和食鹽水分別洗滌;然後用乙酸乙酯萃取,減壓旋幹,柱層析(淋洗劑中乙酸乙酯與石油醚的體積比為1:60)得到淺棕色固體產物。
在100℃的條件下,將中間體(1mmol)與二苄基二硫醚(1.5mmol)加入到10毫升甲苯溶劑中,反應18小時,得到芳基烷基硫醚類衍生物粗品。
將芳基烷基硫醚類衍生物粗品加水洗滌後,用乙酸乙酯萃取,減壓旋幹,柱層析(淋洗劑中乙酸乙酯與石油醚的體積比為1:60)分離可得到純化的目標產物,即芳基烷基硫醚類衍生物,柱層析產率約為78%。
芳基烷基硫醚類衍生物的結構通過1hnmr和13cnmr來測定,具體的檢測圖譜見圖5和圖6。
白色固體產物:
1hnmr(300mhz,cdcl3):δ7.55―7.53(m,2h),7.32―7.27(m,7h),7.12―6.96(m,5h),5.12―5.03(m,2h),4.15(s,2h),3.76(s,2h),3.56(s,2h),1.27―1.25(d,12h,j=6.0hz);
13cnmr(75.5mhz,cdcl3):δ171.0,164.1(jc-f=249.8hz),164.1(jc-f=246.7hz),143.6,142.2,138.6,136.7,133.3,133.1,131.0,130.9,128.9,128.6,127.7,127.4,115.8,115.5,115.0,114.7,95.2,86.5,69.4,59.2,41.2,40.1,37.8,21.6。
實施例4
本實施例提供的一種芳基烷基硫醚類衍生物,其結構式如(iv)式所示。
本實施例還提供一種製備上述芳基烷基硫醚類衍生物的方法:
將丙二酸二乙酯橋連的二炔化合物(20mmol)與對乙基苯乙炔溴(48mmol)加入到反應瓶中,混合在pd(pph3)2cl2和cui作為催化劑的無水無氧催化體系中,其中催化劑的量為0.6mmol,pd(pph3)2cl2:cui=3:1。以三乙胺(100mmol)作鹼,以無水乙腈(40ml)為溶劑,室溫下攪拌反應12小時,將反應產物先後用15%的鹽酸溶液、10%的碳酸氫鈉溶液以及飽和食鹽水分別洗滌;然後用乙酸乙酯萃取,減壓旋幹,柱層析(淋洗劑中乙酸乙酯與石油醚的體積比為1:40)得到淺棕色固體產物。
在95℃的條件下,將中間體(1mmol)與二甲基二硫醚(1.2mmol)加入到7毫升甲苯溶劑中,反應24小時,得到芳基烷基硫醚衍生物粗產品。
將芳基烷基硫醚衍生物粗產品加水洗滌後,用乙酸乙酯萃取,減壓旋幹,柱層析(淋洗劑中乙酸乙酯與石油醚的體積比為1:40)分離可得到純化的目標產物,即芳基烷基硫醚衍生物,柱層析產率約為73%。
芳基烷基硫醚衍生物的結構通過1hnmr和13cnmr以及單晶x射線衍射來測定,具體檢測圖譜見圖7、圖8和圖9。
白色固體產物:
1hnmr(300mhz,cdcl3):δ7.57―7.55(d,2h,j=7.8hz),7.29―7.25(m,4h,j=2.1hz),7.13―7.10(d,2h,j=8.4hz),7.03(s,1h),4.28―4.21(q,4h,j=7.2hz),3.82(s,2h),3.60(s,2h),2.75―2.59(m,4h),2.50(s,3h),1.32―1.19(m,12h);
13cnmr(75.5mhz,cdcl3):δ171.6,144.5,143.6,143.5,143.1,137.8,136.0,134.8,131.3,129.3,127.9,127.5,123.9,120.8,114.4,96.0,86.7,62.0,59.3,41.2,39.9,28.9,28.7,15.7,15.5,14.9,14.1。
綜上所述,本發明實施例1-4提供的一系列新的芳基烷基硫醚類衍生物相對於普通硫醚衍生物,本發明製備的芳基烷基硫醚類衍生物有多環的存在,其結構更加複雜多樣,在化工生產、臨床醫藥中也將表現出更加廣闊的用途前景。本發明的製備方法克服了以往反應中路線過長,底物和反應條件要求苛刻,取代官能團擴展有限的等缺點,該製備方法不但底物合成簡單、試劑比較便宜,並且具有高原子經濟性、綠色環保的得到目標分子,給芳基烷基硫醚類衍生物的工業化生產提供了一條很有價值的途徑。
以上所描述的實施例是本發明一部分實施例,而不是全部的實施例。本發明的實施例的詳細描述並非旨在限制要求保護的本發明的範圍,而是僅僅表示本發明的選定實施例。基於本發明中的實施例,本領域普通技術人員在沒有作出創造性勞動前提下所獲得的所有其他實施例,都屬於本發明保護的範圍。