金屬被覆積層板、其製備方法及其製備軟性電路板的方法與流程
2023-10-19 22:44:47 7
本發明關於一種可應用於軟性印刷電路板的金屬被覆積層板及其製備方法。本發明進一步關於一種使用該金屬被覆積層板製備軟性印刷電路板的方法。
背景技術:
軟性印刷電路板(Flexible Printed Circuit,簡稱:FPC)又稱為可撓性印刷電路板,因具有可撓性及彎曲性,可隨產品可利用的空間大小及形狀進行三度空間的立體配線,加上兼具重量輕、厚度薄的特性,近年來已成為各種高科技設備,如照相機、攝像機、顯示器、磁碟機、印表機及行動電話等產品不可或缺的組件之一。
軟性金屬被覆積層板,例如軟性銅箔基板(Flexible Copper Clad Laminate,簡稱:FCCL)是軟性印刷電路板的上遊材料。現有軟性銅箔基板就構造上可區分為:含接著劑型的三層軟性銅箔基板(3L FCCL)與無接著劑型的二層軟性銅箔基板(2L FCCL)。三層軟性銅箔基板主要是以如環氧樹脂或丙烯酸樹脂的接著劑作為中間層,將銅箔附著於聚醯亞胺層上。二層軟性銅箔基板採用特殊工法製成,不含環氧樹脂或丙烯酸樹脂等耐熱性較低的接著劑,故信賴性較高,且可使產品朝薄型化發展,因此有逐漸取代三層軟性銅箔基板的趨勢。
軟性銅箔基板依產品(印刷電路板)電路配置情形可分為:單面板(single side)及雙面板(double side)。單面板為最基本的軟性銅箔基板,僅在基板的一側具有可供形成電路用的銅箔層,單面板優點包含製程容易、價格較低和良好撓曲性。雙面板是指該軟性銅箔基板上下兩側上均具有銅箔層,因此可在基板的兩面形成電路,且可通過施加導通孔使上下兩側的電路電性連結。因此,雙面板具有更高的集成度、有利於控制電阻,且可兩面同時施作,節省時間;但因厚度增厚,可撓性可能變差。
單面板具有輕薄及優良的撓曲性等優點;雙面板具有優良的機械性和優良的電氣特性(如低的介電常數),且兩面可同時形成電路的優點。然而,上述材料特性與操作性之間往往存在衝突,從事軟性銅箔基板研究的人士常無法兼顧,因此,目前業界仍需一種可同時具備輕薄、優良的可撓曲性、優良的電氣特性且節省後續加工製程時間及成本的軟性銅箔基板。
技術實現要素:
本發明提供一種金屬被覆積層板、其製備方法及其製備軟性電路板的方法,用於解決上述問題。
本發明提供一種金屬被覆積層板,其包含:第一金屬箔;直接設置於該第一金屬箔上的第一聚醯亞胺層;第二金屬箔;及直接設置於該第二金屬箔上的第二聚醯亞胺層;其中該第一聚醯亞胺層接觸於該第二聚醯亞胺層。
在本發明的一實施例中,其中所述第一聚醯亞胺層及第二聚醯亞胺層中至少之一的組成中包含衍生自二氨基矽氧烷單體的聚合單元。
在本發明的一實施例中,其中所述第一聚醯亞胺層及第二聚醯亞胺層的組成中各自包含衍生自二氨基矽氧烷單體的聚合單元。
在本發明的一實施例中,其中所述二氨基矽氧烷單體具有下式(Ⅲ)的通式:
式中,R9各自獨立為H、C1-C4直鏈或支鏈的烷基或苯基;a可相同或不相同且為介於1至6之間的整數;m為介於1至15之間的整數。
在本發明的一實施例中,其中所述式(Ⅲ)中,R9各自獨立為甲基、乙基或苯基;a可相同或不相同且為介於2至5之間的整數;m為介於1至5之間的整數。
在本發明的一實施例中,其中所述二氨基矽氧烷單體選自:
及其組合,
其中m為介於1至5之間的整數。
在本發明的一實施例中,其中所述第一聚醯亞胺層及第二聚醯亞胺層各自具有大於250℃的玻璃化轉換溫度,優選介於260℃至340℃之間的玻璃化轉換溫度。
在本發明的一實施例中,其中所述第一聚醯亞胺層與第一金屬箔、第二聚醯亞胺層與第二金屬箔具有相近或基本相同的熱膨脹係數。
在本發明的一實施例中,其中所述第一金屬箔及第二金屬箔各自具有介於15ppm/℃至25ppm/℃之間的熱膨脹係數。
在本發明的一實施例中,其中所述第一及第二聚醯亞胺層由二酸酐單體與二胺單體發生縮合反應生成的前驅物經醯亞胺化而形成,且所述二氨基矽氧烷單體的用量以所述二胺單體總摩爾數計,為0.1mol%至<10mol%。
在本發明的一實施例中,其中所述二氨基矽氧烷單體的用量以所述二胺單體總摩爾數計,為0.5mol%至7.5mol%。
在本發明的一實施例中,其中所述二氨基矽氧烷單體的用量以所述二胺單體總摩爾數計,為1mol%至<5mol%。
在本發明的一實施例中,其中所述二酸酐單體為芳香族二酸酐單體。
在本發明的一實施例中,其中所述二胺單體包括芳香族二胺單體和所述二氨基矽氧烷單體。
在本發明的一實施例中,其中所述金屬被覆積層板為類雙面二層金屬被覆積層板或雙面二層金屬被覆積層板。
在本發明的一實施例中,其中所述金屬被覆積層板為類雙面二層金屬被 覆積層板,其中所述第一聚醯亞胺層和第二聚醯亞胺層之間具有1gf/cm~500gf/cm的剝離強度。
在本發明的一實施例中,其中所述金屬被覆積層板為類雙面二層金屬被覆積層板,其中所述第一聚醯亞胺層和第二聚醯亞胺層之間具有3gf/cm~100gf/cm的剝離強度。
在本發明的一實施例中,其中所述金屬被覆積層板為類雙面二層金屬被覆積層板,其中所述第一聚醯亞胺層和第二聚醯亞胺層之間具有5gf/cm~50gf/cm的剝離強度。
在本發明的一實施例中,其中所述金屬被覆積層板為雙面二層金屬被覆積層板,其中所述第一聚醯亞胺層和第二聚醯亞胺層之間具有大於500gf/cm的剝離強度。
在本發明的一實施例中,其中所述第一金屬箔及第二金屬箔各自獨立地選自銅箔、鋁箔或銅鋁合金的金屬箔。
本發明提供一種製備上述金屬被覆積層板的方法,包含:
(a)提供一第一金屬膜,所述第一金屬膜包含第一金屬箔及直接設置於所述第一金屬箔上的第一聚醯亞胺層;
(b)提供一第二金屬膜,所述第二金屬膜包含第二金屬箔及直接設置於所述第二金屬箔上的第二聚醯亞胺層;
(c)將第一金屬膜的第一聚醯亞胺層和第二金屬膜的第二聚醯亞胺層重合併進行壓合。
在本發明的一實施例中,其中所述方法的第一聚醯亞胺層及第二聚醯亞胺層各自具有介於260℃至340℃之間的玻璃化轉換溫度。
在本發明的一實施例中,其中步驟(c)控制壓合溫度為300℃~390℃、壓合線壓力為1kgf/cm~60kgf/cm,使第一聚醯亞胺層和第二聚醯亞胺層之間具有1gf/cm~500gf/cm的剝離強度,所述金屬被覆積層板為類雙面二層金屬被覆積層板。
在本發明的一實施例中,其中步驟(c)控制壓合溫度為350℃~400℃、壓合線壓力為100kgf/cm~200kgf/cm,使第一聚醯亞胺層和第二聚醯亞胺層之間具有大於500gf/cm的剝離強度,所述金屬被覆積層板為雙面二層金屬被覆積層板。
本發明提供一種製備軟性電路板的方法,使用前述類雙面二層金屬被覆積層板,且進一步包含以下步驟:
在所述第一金屬箔及所述第二金屬箔的表面上各自形成至少一電路單元;及
將第一聚醯亞胺層和第二聚醯亞胺層分離,形成兩組單面軟性電路板。
在本發明的一實施例中,其中所述製備單面軟性電路板的方法,還包括按照前述的方法製備所述類雙面二層金屬被覆積層板。
本發明提供另一種製備軟性電路板的方法,使用前述雙面二層金屬被覆積層板,且進一步包含以下步驟:
在所述金屬被覆積層板的第一金屬箔及第二金屬箔的表面上各自形成至少一電路單元,製成雙面軟性電路板。
在本發明的一實施例中,其中所述製備雙面軟性電路板的方法,還包括按照前述的方法製備所述雙面二層金屬被覆積層板。
基於上述,本發明所提供的金屬被覆積層板及其製備方法,第一聚醯亞胺層直接設置且附著於該第一金屬箔上,第二聚醯亞胺層直接設置且附著於該第二金屬箔上,而不需在金屬箔與聚醯亞胺層間額外施加接著劑或熱塑性聚醯亞胺(TPI)層以提供接著效果,因此,不但可簡化金屬被覆積層板的製備過程,且所得金屬被覆積層板耐熱性佳,可適用於高溫加工製程,有利於半導體元件的製作。
為使本發明的上述目的、技術特徵及優點能更明顯易懂,下文以部分具體實施例進行詳細說明。
附圖說明
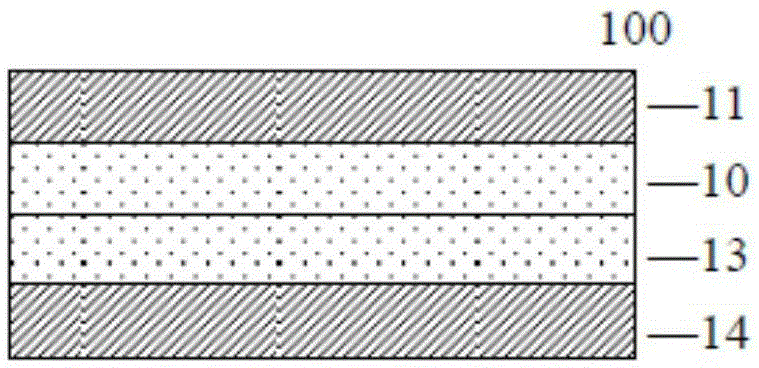
圖1為本發明金屬被覆積層板的示意圖;
圖2為使用本發明金屬被覆積層板製備兩組單面配線軟性電路板的示意圖;
圖3為本發明將兩組單面配線軟性電路板分離的示意圖。
附圖標記說明:
10、20:第一聚醯亞胺層;
11、21:第一金屬箔;
13、23:第二聚醯亞胺層;
14、24:第二金屬箔;
22、25:覆蓋膜;
30、31:滾輪;
100:金屬被覆積層板;
200、210:軟性電路板;
A、B:單面軟性電路板卷。
具體實施方式
為便於理解本文所陳述的揭示內容,現在在下文中定義若干術語。
術語「約」意謂如由一般熟習此項技術者所測定的特定值的可接受誤差,誤差範圍視如何量測或測定該值而定。
圖1為本發明金屬被覆積層板的示意圖;如圖1所示,本發明的金屬被覆積層板100可用於生產軟性印刷電路板,其包含:第一金屬箔11;直接設置於該第一金屬箔11上的第一聚醯亞胺層10;第二金屬箔14;及直接設置於該第二金屬箔14上的第二聚醯亞胺層13。所述第一聚醯亞胺層與第一金屬箔、第二聚醯亞胺層與第二金屬箔具有相近或基本相同的熱膨脹係數。
根據本發明的方案,所述第一金屬箔及第二金屬箔各自為具有介於約15ppm/℃至約25ppm/℃之間的熱膨脹係數的金屬或合金,其例如但不限於:鋁、銅、銀;含鋁、銅、銀中任意組合的合金;或其它具有介於約15ppm/℃至約25ppm/℃之間的熱膨脹係數的合金。根據本發明的較優選實施例,第一金屬箔及第二金屬箔為銅箔、鋁箔、或銅鋁合金的金屬箔。上述銅箔是指銅或以銅為主成分的箔(例如銅含量為90wt%以上的箔),可選自以下群組:延壓退火銅箔(Rolled annealed copper foil,簡稱:Ra銅箔)、電解銅箔(Electrodeposited copper foil,簡稱:ED銅箔)及其組合;上述鋁箔是指鋁或以鋁為主成分的箔(例如鋁含量為90wt%以上的箔);其它金屬箔的定義亦可依此類推。
第一金屬箔及第二金屬箔的厚度並無特殊限制,一般約0.05微米至約50微米之間,較佳介於約0.1微米至約35微米之間,較佳介於約5微米至約20微米之間。
本發明的方案中,第一聚醯亞胺層10直接設置且附著於該第一金屬箔11上,第二聚醯亞胺層13直接設置且附著於該第二金屬箔14上,而不需在金屬箔與聚醯亞胺層間額外施加接著劑或熱塑性聚醯亞胺(TPI)層以提供接著效果,因此,不但可簡化金屬被覆積層板的製備過程,且所得金屬被覆積層板耐熱性佳,可適用於高溫加工製程,有利於半導體元件的製作。
一般用作絕緣支撐層的聚醯亞胺層,必須具有優異的熱安定性,因此會使用結構中具有高對稱性的熱固性聚醯亞胺。但是兩個熱固性聚醯亞胺層彼此附著性很差,所以業界會在一熱固性聚醯亞胺層的表面施加熱塑性聚醯亞胺層(Thermoplastic Polyimide,簡稱:TPI),利用熱塑性聚醯亞胺的特性,加熱使熱塑性聚醯亞胺軟化、熔化,再加壓使其變形並結合至另一熱固性聚醯亞胺層,待溫度下降後,使兩熱固性聚醯亞胺層通過該熱塑性聚醯亞胺層附著在一起。
本發明的另一技術特點在於該第一聚醯亞胺層10直接接觸、附著於該第二聚醯亞胺層13上,而無需額外施加上述的熱塑性聚醯亞胺層。本發明的第一聚醯亞胺層10及第二聚醯亞胺層13中至少一個,較佳為兩者,包含衍生自二氨基矽氧烷的聚合單元,所以該第一聚醯亞胺層10接觸於該第二聚醯亞胺層13時,可產生自粘性(self-adhesive),不需要再使用熱塑性聚醯亞胺,故可以簡化製程。
本發明的方案,對聚醯亞胺層的厚度並無特殊限制,可視原料性質及所需產品特性調整。根據本發明的一實施例,該第一聚醯亞胺層及第二聚醯亞胺層的厚度可各自介於約1微米至約90微米之間,較佳介於約3微米至約50微米之間,更佳介於約5微米至約30微米之間。
本發明的第一聚醯亞胺層及第二聚醯亞胺層是熱固性聚醯亞胺層。如同一般聚醯亞胺的製造方式,所述第一及第二聚醯亞胺層由二酸酐單體與二胺單體發生縮合反應生成的前驅物經醯亞胺化而形成,例如,可使用聚醯胺酸作為前驅物(又稱聚醯亞胺前驅物)而製成。利用聚醯胺酸製備聚醯亞胺的反應流程可簡述如下(其中Ar及Ar'分別為四價有機基團及二價有機基團;n為聚合單元數目):
在上述反應流程所示的製備方法中,是將二胺單體溶於非質子性極性溶劑中,再使其與等摩爾二酸酐單體進行縮合反應形成供製備聚醯亞胺用的前驅物(即聚醯胺酸);接著,以加熱方式使聚醯胺酸進行醯亞胺化(imidization),進一步脫水並環化成聚醯亞胺。
或者,可使用其他聚醯亞胺前驅物或前驅物組合物以製備聚醯亞胺,例如但不限於使用具下式(I)的聚醯亞胺前驅物:
使用包含下述組分的聚醯亞胺前驅物組合物:
及
H2N-P-NH2,
其中G為四價有機基,P為二價有機基且r是0至100的整數(較佳為1至90), Rx各自獨立為H或可感光基,R為有機基。
各種不同聚醯亞胺前驅物的聚合及環化方法以及由其所製得的聚醯亞胺的研發技術已有報導,例如美國專利申請案第11/785,827號、第11/119,555號、第12/846,871號及第12/572,398號與中國專利申請案CN200610162485.X、CN200710138063.3。上述文獻全文併入本文中作為參考。
用以製備上述聚醯亞胺前驅物或前驅物組合物的二酸酐單體一般可為脂肪族或芳香族者,若需耐化性較佳者,較佳為芳香族二酸酐,其實例包括(但不限於)均苯四甲酸二酐(pyromellitic dianhydride,簡稱:PMDA)、3,3',4,4'-聯苯四羧酸二酐(3,3',4,4'-Biphenyltetracarboxylic dianhydride,簡稱:S-BPDA)、4,4'-二酞酸二酐、4,4'-六氟亞異丙基-2,2-雙(酞酸酐)(4,4'-hexafluoroisopropylidene-2,2-bis-(phthalic acid anhydride),簡稱:6FDA)、1-(三氟甲基)-2,3,5,6-苯四羧酸二酐(P3FDA)、二苯甲酮-四羧酸二酐(BTDA)、3,3',4,4'-二苯醚四羧酸二酐(ODPA)、1,4-雙(三氟甲基)-2,3,5,6-苯四羧酸二酐(P6FDA)、1-(3',4'-二羧基苯基)-1,3,3-三甲基茚滿-5,6-二羧酸二酐、1-(3',4'-二羧基苯基)-1,3,3-三甲基茚滿-6,7-二羧酸二酐、1-(3',4'-二羧基苯基)-3-甲基茚滿-5,6-二羧酸二酐、1-(3',4'-二羧基苯基)-3-甲基茚滿-6,7-二羧酸二酐、2,3,9,10-二萘嵌苯四羧酸二酐、1,4,5,8-萘四羧酸二酐、2,6-二氯萘-1,4,5,8-四羧酸二酐、2,7-二氯萘-1,4,5,8-四羧酸二酐、2,3,6,7-四氯萘-2,4,5,8-四羧酸二酐、菲-1,8,9,10-四羧酸二酐、3,3',4,4'-二苯甲酮四羧酸二酐、1,2',3,3'-二苯甲酮四羧酸二酐、3,3',4,4'-二苯甲酮四羧酸二酐、2,2',3,3'-聯苯四羧酸二酐、4,4'-亞異丙基二酞酸二酐、3,3'-亞異丙基二酞酸二酐、4,4'-氧基二酞酸二酐、4,4'-磺醯基二酞酸二酐、3,3'-氧基二酞酸二酐、4,4'-亞甲基二酞酸二酐、4,4'-硫基二酞酸二酐、4,4'-亞乙基二酞酸二酐、2,3,6,7-萘四羧酸二酐、1,2,4,5-萘四羧酸二酐、1,2,5,6-萘四羧酸二酐、苯-1,2,3,4-四羧酸二酐、吡啶-2,3,5,6-四羧酸二酐、1,2,3,4-環丁烷四羧酸二酐(1,2,3,4-cyclobutanetetracarboxylic dianhydride,簡稱:CBDA)、5,5'-(9H-茀-9,9-二基)二異苯並呋喃-1,3-二酮(5,5'-(9H-fluorene-9,9-diyl)diisobenzofuran-1,3-dione,簡稱:BPAF)或其組合。
較佳地,採用選自以下群組的芳香族二酸酐:均苯四酸二酐(PMDA)、3,3',4,4'-聯苯四羧酸二酐(S-BPDA)、4,4'-二酞酸二酐、4,4'-六氟亞異丙基二酞酸二酐(6FDA)、1-(三氟甲基)-2,3,5,6-苯四羧酸二酐(P3FDA)、1,4-雙(三氟甲 基)-2,3,5,6-苯四羧酸二酐(P6FDA)、二苯甲酮-四羧酸二酐(BTDA)、3,3',4,4'-二苯醚四羧酸二酐(ODPA)、1,2,3,4-環丁烷四羧酸二酐(CBDA)、5,5'-(9H-茀-9,9-二基)二異苯並呋喃-1,3-二酮(BPAF)及其組合。
用以製備上述聚醯亞胺前驅物或前驅物組合物的二胺單體一般為芳香族二胺,且可為本領域技術人員所熟知者。舉例來說(但不以此為限)可選自以下群組:4,4'-氧化二苯胺(ODA)、對苯二胺(pPDA)、間二甲基對二氨基聯苯(DMDB)、2,2MD雙(三氟甲基)對聯苯(2,2'-bis(trifluoromethyl)benzidine,TFMB)、鄰二甲基對二氨基聯苯(oTLD)、4,4'-八氟聯苯胺(OFB)、四氟-對-苯二胺(TFPD)、2,2'-5,5'-四氯聯苯胺(TCB)、3,3'-二氯聯苯胺(DCB)、2,2'-雙(3-氨基苯基)六氟丙烷、2,2'-雙(4-氨基苯基)六氟丙烷、4,4'-氧基-雙[3-(三氟甲基)苯胺、3,5-二氨基三氟甲苯(3,5-diaminobenzotrifluoride)、四氟-1,4-苯二胺(tetrafluoro-1,4-phenylene diamine)、四氟-間苯二胺、1,4-雙(4-氨基苯氧基)-2-叔丁基苯(BATB)、2,2'-二甲基-4,4'-雙(4-氨基苯氧基)聯苯(DBAPB)、4,4'-二氨基二環己基甲烷(MDCA)、2,2-雙[4-(4-氨基苯氧基)苯基]六氟丙烷(BAPPH)、2,2'-雙[4-(4-氨基苯氧基)苯基]原冰片烷(BAPN)、5-氨基-1-(4'-氨基苯基)-1,3,3-三甲基茚滿、6-氨基-1-(4'-氨基苯基)-1,3,3-三甲基茚滿、4,4'-亞甲基雙(鄰-氯苯胺)、3,3'-二氯二苯胺、3,3'-磺醯基二苯胺、4,4'-二氨基二苯甲酮、1,5-二氨基萘、雙(4-氨基苯基)二乙基矽烷、雙(4-氨基苯基)二苯基矽烷、雙(4-氨基苯基)乙基膦氧化物、N-(雙(4-氨基苯基))-N-甲基胺、N-(雙(4-氨基苯基))-N-苯基胺、4,4'-亞甲基雙(2-甲基苯胺)、4,4'-亞甲基雙(2-甲氧基苯胺)、5,5'-亞甲基雙(2-氨基苯酚)、4,4'-亞甲基雙(2-甲基苯胺)、4,4'-氧基雙(2-甲氧基苯胺)、4,4'-氧基雙(2-氯苯胺)、2,2'-雙(4-氨基苯酚)、5,5'-氧基雙(2-氨基苯酚)、4,4'-硫基雙(2-甲基苯胺)、4,4'-硫基雙(2-甲氧基苯胺)、4,4'-硫基雙(2-氯苯胺)、4,4'-磺醯基雙(2-甲基苯胺)、4,4'-磺醯基雙(2-乙氧基苯胺)、4,4'-磺醯基雙(2-氯苯胺)、5,5'-磺醯基雙(2-氨基苯酚)、3,3'-二甲基-4,4'-二氨基二苯甲酮、3,3'-二甲氧基-4,4'-二氨基二苯甲酮、3,3'-二氯-4,4'-二氨基二苯甲酮、4,4'-二氨基聯苯、間-苯二胺、4,4'-亞甲基二苯胺(MDA)、4,4'-硫基二苯胺、4,4'-磺醯基二苯胺、4,4'-亞異丙基二苯胺、3,3'-二甲氧基聯苯胺、3,3'-二羧基聯苯胺、2,4-甲苯基二胺、2,5-甲苯基二胺、2,6-甲苯基二胺、間-二甲苯基二胺、2,4-二氨基-5-氯甲苯、2,4-二氨基-6-氯甲苯、1,4-二氨基環己烷(1,4-diaminocyclohexane,CHDA)、 4-(9-(4-氨基苯基)-9H-茀-9-基)苯胺(4-(9-(4-aminophenyl)-9H-fluoren-9-yl)benzenamine,簡稱:BAFL)、9-(4-氨基苯基)-9-苯基-9H-茀基-3-胺(9-(4-aminophenyl)-9-phenyl-9H-fluoren-3-amine)、1-(4-氨基苯基)-2,3-二氫-1,3,3-三甲基-1氫-茚-5-胺(1-(4-aminophenyl)-2,3-dihydro-1,3,3-trimethyl-1H-inden-5-amine,簡稱:TMDA)及其組合。
較佳地,採用4,4'-氧化二苯胺(ODA)、對苯二胺(pPDA)、間二甲基對二氨基聯苯(DMDB)、2,2』-雙(三氟甲基)對聯苯(TFMB)、鄰二甲基對二氨基聯苯(oTLD)、4,4'-亞甲基二苯胺(MDA)、4,4'-二氨基二環己基甲烷(MDCA)、1,4-二氨基環己烷(CHDA)、(4-(9-(4-氨基苯基)-9H-茀-9-基)苯胺(BAFL)、9-(4-氨基苯基)-9-苯基-9H-茀基-3-胺、1-(4-氨基苯基)-2,3-二氫-1,3,3-三甲基-1氫-茚-5-胺(TMDA)或其組合。
為使聚醯亞胺層具有較佳的熱穩定性、機械性質、電氣性與耐化性,更佳地,可採用選自以下群組的芳香族二酸酐與二胺:
二酸酐:均苯四甲酸二酐(PMDA)、3,3',4,4'-聯苯四羧酸二酐(S-BPDA)、4,4'-六氟亞異丙基-2,2-雙(酞酸酐)(6FDA)、二苯甲酮-四羧酸二酐(BTDA)、3,3',4,4'-二苯醚四羧酸二酐(ODPA)及其組合。
二胺:4,4'-氧化二苯胺(ODA)、對苯二胺(pPDA)、間二甲基對二氨基聯苯(DMDB)、2,2』-雙(三氟甲基)對聯苯(2,2'-bis(trifluoromethyl)benzidine,簡稱:TFMB)或其組合。
欲製備具自粘性的聚醯亞胺層,本發明所使用的二胺單體除了上述的芳香族二胺單體外,尚須包含二氨基矽氧烷單體,該二氨基矽氧烷與芳香族二胺一起與二酸酐反應而製得聚醯胺酸。
如上所述,第一聚醯亞胺層10及第二聚醯亞胺層13的組成中包含衍生自二氨基矽氧烷的聚合單元,所以該第一聚醯亞胺層10接觸於該第二聚醯亞胺層13時,可產生自粘性(self-adhesive)。
本發明所使用的二氨基矽氧烷單體並無特殊限制,較佳為具下式(III)的二氨基矽氧烷單體:
其中R9各自獨立為H、C1-C4直鏈或支鏈的烷基、或苯基,較佳為甲基、乙基或苯基,更佳為甲基或苯基;a可相同或不相同且為大於0的整數,較佳為介於1至6之間的整數,更佳介於2至5之間的整數;m為大於0的整數,較佳為介於1至15之間的整數,更佳介於1至5之間的整數。
根據本發明的一實施例,所使用的二氨基矽氧烷單體可為:
或其組合,其中m為介於1至5之間的整數。
該二氨基矽氧烷單體的用量以二胺單體的總摩爾數計,為約0.1mol%至<10mol%,較佳為約0.5mol%至約7.5mol%,更佳為約1mol%至約<5mol%。當二氨基矽氧烷單體含量過多時(例如高於10mol%時),將致使玻璃化轉換溫度(玻璃化溫度)過低,機械強度(如抗拉強度(Tensile Strength)、抗折強度(breaking strength)、尺寸安定性(dimensional stability)、阻燃性能等不佳,且使聚醯亞胺層熱膨脹係數過大,製成積層板後易有翹曲的現象;當二氨基矽氧烷單體含量過低時(例如低於0.1mol%時),則該第一聚醯亞胺層與該第二聚醯亞胺層之間無法產生自粘性。
根據本發明的一實施例,該第一聚醯亞胺層及第二聚醯亞胺層中至少一個具有介於260℃至340℃之間的玻璃化轉換溫度,較佳具有265℃至320℃,更佳為具有270℃至300℃的玻璃化轉換溫度。
根據本發明的一實施例,該第一聚醯亞胺層及第二聚醯亞胺層各自為熱固性聚醯亞胺,具有介於260℃至340℃之間的玻璃化轉換溫度,較佳具有265℃至320℃,更佳為具有270℃至300℃的玻璃化轉換溫度。
在本發明的一較佳具體實施例中,該第一聚醯亞胺層與第一金屬箔、第二聚醯亞胺層與第二金屬箔具有相近或基本相同的熱膨脹係數。較佳該第一聚醯亞胺層及第二聚醯亞胺層各自具有介於15ppm/℃至25ppm/℃之間的熱膨脹係數。第一聚醯亞胺層及第二聚醯亞胺層的熱膨脹係數可隨所選擇的金屬箔做調整;其中,第一聚醯亞胺層及第二聚醯亞胺層的熱膨脹係數可以調整至與第一金屬箔及第二金屬箔的熱膨脹係數相近。例如金屬箔為銅箔時,第一聚醯亞胺層及第二聚醯亞胺層較佳各自具有介於15ppm/℃至19ppm/℃之間的熱膨脹係數。由於該第一聚醯亞胺層及第二聚醯亞胺層的熱膨脹係數與該第一金屬箔及第二金屬箔的熱膨脹係數相近,因此可降低翹曲現象,提升金屬被覆積層的平坦性。
此外,根據本發明的一實施例,本發明的聚醯亞胺可視需要包含含氮雜環官能基,例如但不限於咪唑基、吡啶基、或三唑基,較佳為三唑基。可通過將含氮雜環官能基導入二酸酐單體或二胺單體中製備上述聚醯亞胺,或者在製備聚醯胺酸或聚醯亞胺後(例如通過電漿接枝)將含氮雜環官能基附接至聚合物鏈上。含氮雜環官能基與金屬箔(如銅)可形成錯合物,從而可提升金屬箔與聚醯亞胺的接著度。
本發明的金屬被覆積層板結構上相當於雙面軟性金屬箔(例如銅箔)基板,機械性質較單面軟性銅箔基板優異,且可兩面同時施做電路。但不同於現有雙面軟性銅箔基板的是,本發明可通過調整製備金屬被覆積層板時的壓合溫度和/或壓力,控制第一聚醯亞胺層與第二聚醯亞胺層之間的剝離強度,製備類雙面二層金屬被覆積層板(quasi double-sided 2L metal-clad laminate)或雙面二層金屬被覆積層板。
在本發明的第一具體實施例中,上述類雙面二層金屬被覆積層板中的第一聚醯亞胺層與第二聚醯亞胺層之間具有介於1gf/cm至500gf/cm之間的剝離 強度,較佳為3gf/cm至約100gf/cm的剝離強度,為避免第一聚醯亞胺層與第二聚醯亞胺層之間黏性過大,進行分離時,容易翹曲,更優選第一聚醯亞胺層與第二聚醯亞胺層之間的剝離強度為5gf/cm至約50gf/cm。在此實施例中,該類雙面二層金屬被覆積層板可在該金屬被覆積層板的雙面同時施做電路,製備兩組獨立的軟性印刷電路板,由於該第一聚醯亞胺層和第二聚醯亞胺層的界面具有適當的剝離強度,因此在元件製作完成後,可由該界面進行分離,同時獲得兩組軟性印刷電路板。使用本發明的金屬被覆積層板所製得的軟性印刷電路板具有與利用單面軟性銅箔基板所製得的軟性印刷電路板相當的結構,輕薄、且可撓曲性佳;然而,相較單面軟性銅箔基板而言,使用本發明的類雙面二層金屬被覆積層板可在一次製程中同時製備兩組軟性印刷電路板,產能更為優異,可節省製程時間。此外,一般單面軟性銅箔基板易產生翹曲,因此,在印刷電路時,除了會在銅箔表面施加光阻劑用於電路製作之外,也會在聚醯亞胺層表面施加光阻劑以使軟性銅箔基板兩對側結構平衡,減少翹曲發生,此光阻劑會在後續步驟除去,然而,此作法徒增製程成本。本發明的類雙面二層金屬被覆積層板本身結構對稱且可同時施作電路,因此,相對於一般單面軟性銅箔基板而言,不但不易發生翹曲,且可以較快速且經濟的方式製成軟性印刷電路板。
在本發明的第二具體實施例中,上述雙面二層金屬被覆積層板中的第一聚醯亞胺層與第二聚醯亞胺層之間具有大於500gf/cm的剝離強度,較佳為具有大於800gf/cm,更佳為大於1000gf/cm的剝離強度。在此實施例中,該第一聚醯亞胺層和第二聚醯亞胺層的界面剝離強度大,粘著性佳,因此可作為雙面金屬被覆積層板,供製備需雙面配線的軟性印刷電路板用。
本發明另提供一種製備上述金屬被覆積層板的方法。本發明的方法包含下列步驟:
(a)提供一第一金屬膜,該第一金屬膜包含第一金屬箔及直接設置於該第一金屬箔上的第一聚醯亞胺層;
(b)提供一第二金屬膜,該第二金屬膜包含第二金屬箔及直接設置於該第二金屬箔上的第二聚醯亞胺層;及
(c)將第一金屬膜的第一聚醯亞胺層和第二金屬膜的第二聚醯亞胺層重合併進行壓合,
其中該第一金屬箔及第二金屬箔各自具有介於15ppm/℃至25ppm/℃之間的熱膨脹係數。
該第一金屬箔、該第二金屬箔、該第一聚醯亞胺層及該第二聚醯亞胺層的材料及性質如本文先前所描述。
上述步驟(a)及(b)中,該第一金屬膜及第二金屬膜各自為無接著劑的二層軟性金屬膜。製備該第一金屬膜及第二金屬膜的方法並無特殊限制,例如,可為濺鍍/電鍍法(sputtering/plating)、塗布法(casting)或熱壓合法(lamination)。舉例言之,第一、濺鍍/電鍍法:在高真空的環境下,以濺鍍的方式沉積一層金屬薄膜(約在1微米以下)在聚醯亞胺膜上,以微影蝕刻的方式粗化表面,再以電鍍的方式將金屬層增厚至所需厚度;第二、塗布法:以金屬箔為基材,先塗上一層薄的熱塑性聚醯亞胺前驅物,經乾燥後,再塗上第二層較厚的聚醯亞胺前驅物(一般是熱固性)以增加基板的剛性,經高溫環化後形成兩層式軟板;第三、熱壓合法:以聚醯亞胺膜為基材,先塗上一層薄的熱塑性聚醯亞胺前驅物,經高溫環化後將金屬箔放置在熱塑性聚醯亞胺上,在氮氣下以高溫熱滾輪的方式,在適當的貼合壓力下,將熱塑性聚醯亞胺重新熔融與金屬箔壓合,形成兩層式軟板。
根據本發明的一實施例,可先將芳香族二胺單體及二氨基矽氧烷單體與芳香族二酸酐反應製備聚醯胺酸溶液(例如但不限於在0℃至80℃下反應1至48小時),將該聚醯胺酸溶液塗布在金屬箔上(塗布厚度例如但不限約2微米至180微米)後進行預烤以移除溶劑(例如但不限於在50℃至200℃的溫度下加熱1分鐘至20分鐘),接著再進一步加熱使該聚醯胺酸進行脫水、環化成聚醯亞胺(例如但不限於在250℃至350℃的溫度下加熱30分鐘至180分鐘)。
根據本發明的另一實施例,可使用玻璃或塑膠為基材,將聚醯亞胺前驅物或前驅物組合物塗布於基材,形成一包含基材和樹脂層的半成品,將該半成品加熱乾燥以移除溶劑,形成一包含基材和樹脂層的成品,將上述的成品的樹脂層表面,以上述濺鍍/電鍍法或熱壓合法,在樹脂層上形成金屬箔層,隨後移除該玻璃或塑膠基材並進一步加熱,形成兩層式軟板。上述塑膠基材較佳為聚對苯二甲酸乙二酯、聚甲基丙烯酸甲酯、聚環烯烴樹脂、三醋酸纖維素或其混合物。上述加熱乾燥所移除的溶劑是指製備本發明聚醯亞胺前驅物時為溶解單體或其他目的所添加的溶劑,其種類並無任何特別的限制,較 佳為極性非質子性溶劑。舉例言之(但不以此為限),該非質子性溶劑可選自以下群組:N,N-二甲基乙醯胺(DMAc)、N-甲基吡咯烷酮(NMP)、N,N-二甲基甲醯胺(DMF)、四甲基脲(tetramethylurea,簡稱:TMU)、二甲基亞碸(Dimethylsulfoxide,簡稱:DMSO)、及其組合,較佳為N,N-二甲基乙醯胺(DMAc)。
上述步驟(c)中,該第一聚醯亞胺層及第二聚醯亞胺層間無接著劑。該步驟(c)可選任何方式,較佳以卷對卷(roll to roll)方式進行,將第一金屬膜的第一聚醯亞胺層朝向第二金屬膜的第二聚醯亞胺層,然後進行重合。上述步驟(c)可選任何方式進行壓合,例如但不限於:滾輪壓合(roller lamination)、熱板壓合(hot press)、真空壓合(vacuum lamination)或真空快壓(vacuum press),較佳為滾輪壓合。視需要,可施加保護膜在金屬膜上(保護膜/第一金屬膜或第二金屬膜/保護膜)一起進行壓合,上述保護膜的種類並無特殊限制,例如,可採用日本鍾淵(KANEKA)化學公司的NPI作為保護膜。
上述步驟(c)所用的聚醯亞胺層是由二酸酐單體與二胺單體進行開環聚合成聚醯胺酸後,再經醯亞胺化(imidization)脫水而成聚醯亞胺。所得聚醯亞胺結構擁有高對稱性,玻璃化轉換溫度介於260℃至340℃之間,具有優異的熱安定性;且因為具有與金屬箔相近的熱膨脹係數,所以可避免產生翹曲。
另,本發明的第一聚醯亞胺層及第二聚醯亞胺層中至少一個包含衍生自二氨基矽氧烷的聚合單元,較佳為該第一聚醯亞胺層及該第二聚醯亞胺層均包含衍生自二氨基矽氧烷的聚合單元,所以該第一聚醯亞胺層與該第二聚醯亞胺層壓合後,可產生自粘性(self-adhesive)。舉例言之,將該第一聚醯亞胺層與該第二聚醯亞胺層重合後,升高溫度及壓力後用輥壓機進行壓合,可使該第一聚醯亞胺層與該第二聚醯亞胺層之間產生化學鍵結(包括氫鍵結和共價鍵結),藉此增加接著力。上述的溫度及壓力取決於第一聚醯亞胺層與第二聚醯亞胺層之間所需的剝離強度。
上述步驟(c)的壓合較佳是在大於該第一聚醯亞胺層及該第二聚醯亞胺層的玻璃化轉換溫度的溫度下進行。壓合的溫度及壓力可視所欲制的產品進行調整。本案發明人經過反覆實驗及研究後發現:可通過壓合的溫度及壓力與該第一聚醯亞胺層及該第二聚醯亞胺層的玻璃化轉換溫度搭配,製備類雙面二層金屬被覆積層板或雙面二層金屬被覆積層板。
根據本發明的一具體實施例,為避免翹曲,選滾輪壓合方式搭配高溫低壓進行壓合,該第一聚醯亞胺層及該第二聚醯亞胺層的玻璃化轉換溫度介於260℃~340℃之間,控制壓合溫度介於300℃~390℃之間,壓合線壓力介於1kgf/cm~60kgf/cm之間,較優選為介於5kgf/cm~50kgf/cm之間,所得金屬被覆積層板是類雙面二層金屬被覆積層板,該第一聚醯亞胺層及該第二聚醯亞胺層的界面的剝離強度介於1gf/cm~500gf/cm之間。根據本發明的具體實施例,類雙面二層金屬被覆積層板的剝離強度可為3、5、6、7、8、10、15、30、45、60、75、90、100、130、150、200、300、400或500gf/cm。根據本發明的一較佳實施例,使用輥壓機滾輪壓合該第一聚醯亞胺層及該第二聚醯亞胺層,壓合溫度較佳為介於310℃~370℃之間,線壓力較佳為介於10kgf/cm~45kgf/cm之間,所得金屬被覆積層板是類雙面二層金屬被覆積層板,該第一聚醯亞胺層及該第二聚醯亞胺層的界面的剝離強度較佳介於3gf/cm~100gf/cm之間,更佳介於5gf/cm~50gf/cm之間。在上述壓合條件下所形成的類雙面二層金屬被覆積層板,第一聚醯亞胺層和第二聚醯亞胺層間具有適當的附著性,可通過軟性電路板的相關製程完成軟性電路板的製作,當製成軟性電路板後,又可輕易自該第一聚醯亞胺層與該第二聚醯亞胺層之間分離,成為兩單面軟性電路板。上述的線壓力:是指在一組滾輪式的熱壓機設備中,在固定寬幅的基材上,兩滾輪對於基材施以一固定的力量做壓合,將此力量除上基材的寬度,即為壓合的線壓力。
根據本發明的另一具體實施例,該第一聚醯亞胺層及該第二聚醯亞胺層的玻璃化轉換溫度介於260℃~340℃之間,通過調整壓合溫度及壓力,本發明的方法也可製備雙面二層金屬被覆積層板。例如,選滾輪壓合方式進行壓合,使用介於350℃~400℃之間的壓合溫度及介於100kgf/cm~200kgf/cm之間的壓合線壓力,使該第一聚醯亞胺層及第二聚醯亞胺層的界面產生大於500gf/cm的剝離強度,較佳為大於800gf/cm,更佳為大於1000gf/cm,該第一聚醯亞胺層及該第二聚醯亞胺層即可有效附著在一起,而不分離。
一般在製造單面軟性電路板時,為防止在製程中產生翹曲,通常會在單面銅箔基板金屬的上下表面均貼附幹膜光阻,造成光阻的浪費。為節省製程時間,另有業者將兩個單面銅箔基板的聚醯亞胺層以膠帶進行貼合,並在兩面施作電路後,再將其分離。然而,以膠帶進行貼合的方法一般僅適用於板 對板(sheet by sheet)的製程,應用至卷對卷(roll to roll)製程時有其困難度,無法以卷對卷製程連續且快速地生產;此外,由於這類膠帶大多為環氧樹脂或丙烯酸酯等不耐高溫,且耐化性不佳,而印刷電路板製作一般包含酸性電鍍、酸性蝕刻及鹼性顯影、鍍金、化鎳浸金(electroless nickel immersion gold,簡稱ENIG)等製程,往往必須在膠帶失效後(例如,在蝕刻後),將其移除,以新膠帶重新貼合後再進行後續製程,因此不但製程繁複且可能有殘膠遺留。本發明的金屬被覆積層板的製造方法則無上述缺點,更適用於卷對卷製程。另外,現有技術中製造雙面軟性電路板時,因為熱固性聚醯亞胺層之間,彼此附著性很差(一般而言,剝離強度約<1gf/cm),為讓熱固性聚醯亞胺層之間具有附著力,一般常使用熱塑性聚醯亞胺,舉例言之,臺灣專利申請案第200709751A號揭露用熱塑性聚醯亞胺粘合兩聚醯亞胺層,但是這增加了製程的複雜性。再者,熱塑性聚醯亞胺常通過導入柔性基團(例如C=O、─O─、─S─)以降低主鏈剛硬性、導入不對稱結構單體來降低高分子對稱性、使用非平面結構單體來降低共平面結構,也可以通過降低規則性而降低玻璃化轉換溫度。所以一般而言,熱塑性聚醯亞胺的玻璃化轉換溫度(Tg)較低(約170℃~250℃),熱膨脹係數較高(約40ppm/℃~90ppm/℃),容易造成積層板翹曲。此外,由於熱塑性聚醯亞胺的玻璃化轉換溫度較低,也不利此雙面式基板的耐熱性。
因此,本發明的方法可通過調整適當的壓合溫度與壓力,製備類雙面二層金屬被覆積層板,且可在類雙面二層金屬被覆積層板的雙面製備電路後,輕易將其分離成兩單面軟性電路板,故可解決目前業界需在單面銅箔基板金屬的上下表面均貼附幹膜光阻或以膠帶貼合製造單面軟性電路板的缺點,具有簡化製程和節省成本的優點。另,本發明也可調整適當的壓合溫度與壓力,製備雙面二層金屬被覆積層板,可解決業界使用熱塑型聚醯亞胺製造雙面金屬被覆積層板的缺點,不僅節省成本更增加了基板的耐熱性。
本發明的金屬被覆積層板可用於製備單面或雙面的軟性電路板。本發明的金屬被覆積層板因不含接著劑或在金屬箔及聚醯亞胺層間提供接著力用的熱塑性聚醯亞胺層,因此,可製得輕且薄的軟性電路板。此外,由於聚醯亞胺層與金屬箔具有相近的熱膨脹係數,可減少翹曲。
因此,本發明進一步提供一種製備單面的軟性電路板的方法,使用前述類雙面二層金屬被覆積層板,且進一步包含以下步驟:
(d)在所述金屬被覆積層板的第一金屬箔及第二金屬箔的表面上各自形成至少一電路單元;及
(e)將第一聚醯亞胺層和第二聚醯亞胺層分離,形成兩組單面軟性電路板。
本領域技術人員可理解,上述步驟(d)中形成電路單元用的第一金屬箔表面是指該第一金屬箔中相對於該第一金屬箔與該第一聚醯亞胺層附著表面的面,形成電路單元用的第二金屬箔表面是指該第二金屬箔中相對於該第二金屬箔與該第二聚醯亞胺層附著表面的面。
上述步驟(d)中形成電路單元的方法並無特殊限制,可使用本領域技術人員所現有適當方法,例如,如圖2所示(圖2為使用本發明金屬被覆積層板製備兩組單面配線軟性電路板的示意圖),可通過曝光、顯影、蝕刻及移除光阻劑等步驟,將位於第一聚醯亞胺層20上的第一金屬箔21及位於第二聚醯亞胺層23上的第二金屬箔24各自圖案化以製備獨立的電路單元,隨後可視需要在經圖案化的該第一金屬箔21和/或第二金屬箔24上施加覆蓋膜(coverlay)22及覆蓋膜25以保護電路單元,並視需要進行化鎳浸金製程(ENIG)(圖上未示出)。隨後在步驟(e)中,自該第一聚醯亞胺層20及第二聚醯亞胺層23的界面進行分離,形成兩組單面配線軟性電路板200及軟性電路板210(參考圖2)。
由於該第一聚醯亞胺層及該第二聚醯亞胺層的界面具有適當但不過度的剝離強度(介於1gf/cm~500gf/cm之間),因此,在步驟(e)中,可利用例如捲軸式分離法,自該界面通過滾輪30及滾輪31進行分離,形成兩組單面軟性電路板200及軟性電路板210,並分別收成單面軟性電路板卷A及單面軟性電路板卷B(參考圖3,圖3為本發明將兩組單面配線軟性電路板分離的示意圖)。
本領域技術人員可理解,本發明的金屬被覆積層板因具有雙面金屬箔,故除可用於製備單面軟性電路板,也可以用於製備雙面軟性電路板,尤其當第一聚醯亞胺層及第二聚醯亞胺層的界面具有大於500gf/cm的剝離強度時。
因此,本發明還進一步提供一種製備雙面的軟性電路板的方法,使用前述雙面二層金屬被覆積層板,且進一步包含以下步驟:
(f)在所述金屬被覆積層板的第一金屬箔及第二金屬箔的表面上各自形成 至少一電路單元;
上述步驟(f)中形成電路單元的方法如步驟(d)所述。可使用本領域技術人員所現有適當方法,例如但不限於,在上述步驟((d)之後,進一步蝕刻所暴露出來的第一聚醯亞胺層及第二聚醯亞胺層形成一通孔在該通孔中濺鍍種子層後再鍍覆導電元件,電性連結上、下線路層。
鑑於此,本發明的金屬被覆積層板不但兼具單面板輕、薄的優點及雙面板可同時製作雙面線路的優點,且可用以製備單面軟性電路板或雙面軟性電路板,因此,相較於現有單面軟性銅箔基板或雙面軟性銅箔基板用途更為廣泛。此外,本發明的金屬被覆積層板製備方式簡單,成本便宜,也具經濟優勢。
本發明的較佳實施方式已揭露如上,但是其是用於對本發明作進一步說明,而非用以限制本發明的範圍。任何本領域技術人員可輕易達成的修飾及改變均包括於本案說明書揭示內容及所附申請專利範圍的範圍內。
實施例
以下實施例中所提及的縮寫定義如下:
PAN-H:m=1
PAN-P:m=1
製備例1
將218.12克(1摩爾)的均苯四酸二酐(pyromellitic dianhydride,簡稱:PMDA)溶於1291克的NMP中,加熱所得混合物至50℃且反應攪拌兩個小時。慢慢滴入11.62克(0.1摩爾)的丙烯酸2-羥基乙酯(2-hydroxyethyl acrylate,簡稱:HEA),在50℃下反應攪拌兩個小時。其後,將199.24克(0.995摩爾)的ODA及1.24克(0.005摩爾)的PAN-H加至溶液中,待完全溶解後,在50℃下反應攪拌 六個小時,可得聚醯亞胺前驅樹脂PAA-1,固含量為25%,PAN-H約佔二胺單體總摩爾數的0.5mol%。
製備例2
將218.12克(1摩爾)的PMDA溶於1293克的NMP中,加熱所得混合物至50℃且反應攪拌兩個小時。慢慢滴入11.62克(0.1摩爾)的HEA,在50℃下反應攪拌兩個小時。其後,將196.24克(0.98摩爾)的ODA及4.97克(0.02摩爾)的PAN-H加至溶液中,待完全溶解後,在50℃下反應攪拌六個小時,可得聚醯亞胺前驅樹脂PAA-2,固含量為25%,PAN-H約佔二胺單體總摩爾數的2mol%。
製備例3
將218.12克(1摩爾)的PMDA溶於1297克的N-甲基吡咯烷酮(N-methyl-2-pyrrolidone,簡稱:NMP)中,加熱所得混合物至50℃且反應攪拌兩個小時。慢慢滴入11.62克(0.1摩爾)的HEA,在50℃下反應攪拌兩個小時。其後,將190.43克(0.951摩爾)的4,4'-氧化二苯胺(4,4'-oxydianiline,簡稱:ODA)及12.18克(0.049摩爾)的PAN-H加至溶液中,待完全溶解後,在50℃下反應攪拌六個小時,可得聚醯亞胺前驅樹脂PAA-3,固含量為25%,PAN-H約佔二胺單體總摩爾數的4.9mol%。
製備例4
將218.12克(1摩爾)的PMDA溶於1300克的NMP中,加熱所得混合物至50℃且反應攪拌兩個小時。慢慢滴入11.62克(0.1摩爾)的HEA,在50℃下反應攪拌兩個小時。其後,將186.22克(0.93摩爾)的ODA及17.40克(0.07摩爾)的PAN-H加至溶液中,待完全溶解後,在50℃下反應攪拌六個小時,可得聚醯亞胺前驅樹脂PAA-4,固含量為25%,PAN-H約佔二胺單體總摩爾數的7mol%。
製備例5
將218.12克(1摩爾)的PMDA溶於1304克的NMP中,加熱所得混合物至50℃且反應攪拌兩個小時。慢慢滴入11.62克(0.1摩爾)的HEA,在50℃下反應攪拌兩個小時。其後,將180.22克(0.9摩爾)的ODA及24.85克(0.1摩爾)的PAN-H加至溶液中,待完全溶解後,在50℃下反應攪拌六個小時,可得聚醯亞胺前驅樹脂PAA-5,固含量為25%,PAN-H約佔二胺單體總摩爾數的10mol%。
製備例6
將218.12克(1摩爾)的PMDA溶於1334克的NMP中,加熱所得混合物至50 ℃且反應攪拌兩個小時。慢慢滴入11.62克(0.1摩爾)的HEA,在50℃下反應攪拌兩個小時。其後,將190.43克(0.951摩爾)的ODA及24.34克(0.049摩爾)的PAN-P加至溶液中,待完全溶解後,在50℃下反應攪拌六個小時,可得聚醯亞胺前驅樹脂PAA-6,固含量為25%,PAN-P約佔二胺單體總摩爾數的4.9mol%。
製備例7
將218.12克(1摩爾)的PMDA溶於1290克的NMP中,加熱所得混合物至50℃且反應攪拌兩個小時。慢慢滴入11.62克(0.1摩爾)的HEA,在50℃下反應攪拌兩個小時。其後,將200.24克(1摩爾)的ODA加至溶液中,待完全溶解後,在50℃下反應攪拌六個小時,可得聚醯亞胺前驅樹脂PAA-7,固含量為25%,PAN-H約佔二胺單體總摩爾數的0mol%。
製備例8
將218.12克(1摩爾)的PMDA溶於1307克的NMP中,加熱所得混合物至50℃且反應攪拌兩個小時。慢慢滴入11.62克(0.1摩爾)的HEA,在50℃下反應攪拌兩個小時。其後,將176.21克(0.88摩爾)的ODA及29.82克(0.12摩爾)的PAN-H加至溶液中,待完全溶解後,在50℃下反應攪拌六個小時,可得聚醯亞胺前驅樹脂PAA-8,固含量為25%,PAN-H約佔二胺單體總摩爾數的12mol%。
製備例B1
將294.22克(1摩爾)的3,3,4,4-聯苯四甲酸二酐(4,4'-Biphthalic dianhydride,簡稱:BPDA)溶於1298克的NMP中,加熱所得混合物至50℃且反應攪拌兩個小時。慢慢滴入11.62克(0.1摩爾)的HEA,在50℃下反應攪拌兩個小時。其後,將86.51克(0.8摩爾)的對苯二胺(p-Phenylenediamine,簡稱:PPDA)、39.05克(0.195摩爾)的ODA及1.24克(0.005摩爾)的PAN-H加至溶液中,待完全溶解後,在50℃下反應攪拌六個小時,可得聚醯亞胺前驅樹脂PAA-B1,固含量為25%,PAN-H約佔二胺單體總摩爾數的0.5mol%。
製備例B2
將294.22克(1摩爾)的BPDA溶於1300克的NMP中,加熱所得混合物至50℃且反應攪拌兩個小時。慢慢滴入11.62克(0.1摩爾)的HEA,在50℃下反應攪 拌兩個小時。其後,將86.51克(0.8摩爾)的PPDA、36.04克(0.18摩爾)的ODA及4.97克(0.02摩爾)的PAN-H加至溶液中,待完全溶解後,在50℃下反應攪拌六個小時,可得聚醯亞胺前驅樹脂PAA-B2,固含量為25%,PAN-H約佔二胺單體總摩爾數的2mol%。
製備例B3
將294.22克(1摩爾)的BPDA溶於1304克的NMP中,加熱所得混合物至50℃且反應攪拌兩個小時。慢慢滴入11.62克(0.1摩爾)的HEA,在50℃下反應攪拌兩個小時。其後,將86.51克(0.8摩爾)的PPDA、30.24克(0.151摩爾)的ODA及12.18克(0.049摩爾)的PAN-H加至溶液中,待完全溶解後,在50℃下反應攪拌六個小時,可得聚醯亞胺前驅樹脂PAA-B3,固含量為25%,PAN-H約佔二胺單體總摩爾數的4.9mol%。
製備例B4
將294.22克(1摩爾)的BPDA溶於1307克的NMP中,加熱所得混合物至50℃且反應攪拌兩個小時。慢慢滴入11.62克(0.1摩爾)的HEA,在50℃下反應攪拌兩個小時。其後,將86.51克(0.8摩爾)的PPDA、26.03克(0.13摩爾)的ODA及17.40克(0.07摩爾)的PAN-H加至溶液中,待完全溶解後,在50℃下反應攪拌六個小時,可得聚醯亞胺前驅樹脂PAA-B4,固含量為25%,PAN-H約佔二胺單體總摩爾數的7mol%。
製備例B5
將294.22克(1摩爾)的BPDA溶於1312克的NMP中,加熱所得混合物至50℃且反應攪拌兩個小時。慢慢滴入11.62克(0.1摩爾)的HEA,在50℃下反應攪拌兩個小時。其後,將86.51克(0.8摩爾)的PPDA、20.02克(0.1摩爾)的ODA及24.85克(0.1摩爾)的PAN-H加至溶液中,待完全溶解後,在50℃下反應攪拌六個小時,可得聚醯亞胺前驅樹脂PAA-B5,固含量為25%,PAN-H約佔二胺單體總摩爾數的10mol%。
製備例B6
將294.22克(1摩爾)的BPDA溶於1341克的NMP中,加熱所得混合物至50℃且反應攪拌兩個小時。慢慢滴入11.62克(0.1摩爾)的HEA,在50℃下反應攪拌兩個小時。其後,將86.51克(0.8摩爾)的PPDA、30.24克(0.151摩爾)的ODA及24.34克(0.049摩爾)的PAN-P加至溶液中,待完全溶解後,在50℃下反應攪拌 六個小時,可得聚醯亞胺前驅樹脂PAA-B6,固含量為25%,PAN-P約佔二胺單體總摩爾數的4.9mol%。
製備例B7
將294.22克(1摩爾)的BPDA溶於1297克的NMP中,加熱所得混合物至50℃且反應攪拌兩個小時。慢慢滴入11.62克(0.1摩爾)的HEA,在50℃下反應攪拌兩個小時。其後,將86.51克(0.8摩爾)的PPDA、40.05克(0.2摩爾)的ODA加至溶液中,待完全溶解後,在50℃下反應攪拌六個小時,可得聚醯亞胺前驅樹脂PAA-B7,固含量為25%,PAN-H約佔二胺單體總摩爾數的0mol%。
製備例B8
將294.22克(1摩爾)的BPDA溶於1315克的NMP中,加熱所得混合物至50℃且反應攪拌兩個小時。慢慢滴入11.62克(0.1摩爾)的HEA,在50℃下反應攪拌兩個小時。其後,將86.51克(0.8摩爾)的PPDA、16.02克(0.08摩爾)的ODA及29.82克(0.12摩爾)的PAN-H加至溶液中,待完全溶解後,在50℃下反應攪拌六個小時,可得聚醯亞胺前驅樹脂PAA-B8,固含量為25%,PAN-H約佔二胺單體總摩爾數的12mol%。
實施例1(類雙面二層金屬被覆積層板)
將製備例1中合成的聚醯亞胺前驅樹脂溶液PAA-1以滾輪均勻塗布於銅箔上(長春VLP銅箔,1/3oz(12μm)),以120℃烘烤5分鐘,接著以350℃氮氣烘箱烘烤120分鐘,即可得到具有聚醯亞胺塗層的單面銅箔積層板,聚醯亞胺塗層厚度約12μm。
取兩組上述的單面銅箔積層板,以聚醯亞胺層為內層,銅箔為外層,將其重合併以熱滾輪壓合,壓合條件為線壓力20kgf/cm、壓合溫度380℃,之後冷卻可得本發明的類雙面二層金屬被覆積層板Cu-PI-1。
上述的線壓力:是指在一組滾輪式的熱壓機設備中,在固定寬幅的基材上,兩滾輪對於基材施以一固定的力量做壓合,將此力量除上基材的寬度,即為壓合的線壓力。
實施例2(雙面二層金屬被覆積層板)
同實施例1的方法,唯將壓合條件改為線壓力190kgf/cm、壓合溫度400℃, 冷卻後可得本發明的金屬被覆積層板Cu-PI-2。
實施例3(類雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-2,壓合條件改為線壓力20kgf/cm、壓合溫度360℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-3。
實施例4(雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-2,將壓合條件改為線壓力140kgf/cm、壓合溫度390℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-4。
實施例5(類雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-3,壓合條件改為線壓力20kgf/cm、壓合溫度360℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-5。
實施例6(類雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-3,壓合條件改為線壓力60kgf/cm、壓合溫度320℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-6。
實施例7(雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-3,壓合條件改為線壓力190kgf/cm、壓合溫度350℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-7。
實施例8(雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-3,壓合條件改為線壓力140kgf/cm、壓合溫度390℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-8。
實施例9(類雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-4,壓合條件改為線壓力20kgf/cm、壓合溫度340℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-9。
實施例10(雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-4,將壓合條件改為線壓力120kgf/cm、壓合溫度390℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-10。
實施例11(類雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-5,壓合條件改為線壓力20kgf/cm、壓合溫度330℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-11。
實施例12(雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-5,將壓合條件改為線壓力110kgf/cm、壓合溫度390℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-12。
實施例13(類雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-6,壓合條件改為線壓力20kgf/cm、壓合溫度370℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-13。
實施例14(類雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-6,壓合條件改為線壓力60kgf/cm、壓合溫度320℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-14。
實施例15(雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-6,將壓合條件改為線壓力140kgf/cm、壓合溫度390℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-15。
比較例16
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-7,將壓合條件改為線壓力140kgf/cm、壓合溫度390℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-16。
比較例17
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-8,將壓合條件改為線壓力20kg/cm、壓合溫度330℃,冷卻後可得本發明的金屬被覆積層板 Cu-PI-17。
比較例18
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-8,將壓合條件改為線壓力110kgf/cm、壓合溫度390℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-18。
實施例B1(類雙面二層金屬被覆積層板)
實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-B1,壓合條件改為線壓力20kgf/cm、壓合溫度380℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-b1。
實施例B2(雙面二層金屬被覆積層板)
實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-B1,壓合條件改為線壓力190kgf/cm、壓合溫度400℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-b2。
實施例B3(類雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-B2,壓合條件改為線壓力20kgf/cm、壓合溫度370℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-b3。
實施例B4(雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-B2,將壓合條件改為線壓力140kgf/cm、壓合溫度390℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-b4。
實施例B5(類雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-B3,壓合條件改為線壓力20kgf/cm、壓合溫度370℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-b5。
實施例B6(類雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-B3,壓合條件改為線壓力60kgf/cm、壓合溫度320℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-b6。
實施例B7(雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-B3,壓合條件改為線壓力190kgf/cm、壓合溫度350℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-b7。
實施例B8(雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-B3,壓合條件改為線壓力140kgf/cm、壓合溫度390℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-b8。
實施例B9(類雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-B4,壓合條件改為線壓力20kgf/cm、壓合溫度340℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-b9。
實施例B10(雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-B4,將壓合條件改為線壓力120kgf/cm、壓合溫度390℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-b10。
實施例B11(類雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-B5,壓合條件改為線壓力20kgf/cm、壓合溫度330℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-b11。
實施例B12(雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-B5,將壓合條件改為線壓力110kgf/cm、壓合溫度390℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-b12。
實施例B13(類雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-B6,壓合條件改為線壓力20kgf/cm、壓合溫度370℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-b13。
實施例B14(類雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-B6,壓合條件改為線壓力60kgf/cm、壓合溫度320℃,冷卻後可得本發明的金屬被覆積層板 Cu-PI-b14。
實施例B15(雙面二層金屬被覆積層板)
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-B6,將壓合條件改為線壓力140kgf/cm、壓合溫度390℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-b15。
比較例B16
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-B7,將壓合條件相同:線壓力140kgf/cm、壓合溫度390℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-b16。
比較例B17
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-B8,將壓合條件改為線壓力20kg/cm、壓合溫度330℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-b17。
比較例B18
同實施例1的方法,唯將聚醯亞胺前驅樹脂溶液改為PAA-B8,將壓合條件改為線壓力110kg/cm、壓合溫度390℃,冷卻後可得本發明的金屬被覆積層板Cu-PI-b18。
聚醯亞胺層玻璃化轉換溫度(Tg)量測:
將單面金屬被覆積層板的聚醯亞胺層取下,以熱機械分析儀(TMA,德州儀器公司的TA Q400儀器)量測Tg數據。量測範圍為0至500℃,升溫速度為10℃/分。
聚醯亞胺層熱膨脹係數(CTE)量測:
將單面金屬被覆積層板的聚醯亞胺層取下,以熱機械分析儀(TMA,德州儀器公司的TA Q400儀器)量測CTE數據。量測範圍為0至500℃,升溫速度為10℃/分。
兩聚醯亞胺層間的剝離強度量測:
將上述實施例與比較例壓合完成的積層板裁切成15cm×1cm的測試條,將測試條末端處的兩聚醯亞胺層些微分開,分別夾於微電腦拉力測試機 (HT-9102,弘達公司,最高荷重100公斤)的兩夾具頭;且在兩夾具頭相距1cm下,以上下180度對拉方式進行剝離強度測試。
抗張強度量測:
抗張強度(tensile strength)是依據IPC-TM-650(2.4.19)方法,使用設備為萬能拉力機,測量上述實施例與比較例的壓合前的單面銅箔積層板,去除銅箔之後的聚醯亞胺膜的機械特性。高於100Mpa為合格。
阻燃測試:
阻燃測試是根據UL94標準針對聚醯亞胺膜進行測試。
各實施例與比較例相關測試結果如表1~4所示:
以實施例1至15與實施例B1至B15兩組實驗的結果顯示,通過調整壓合溫度及壓力可製得具適當剝離強度的類雙面二層金屬被覆積層板或高剝離強度的雙面二層金屬積層板。此外,結果也顯示實施例1至15與實施例B1至B15的金屬被覆積層板具有與銅箔相近的熱膨脹係數,且抗翹曲及抗張強度方面均符合要求。
由比較例16及比較例B16(未使用二氨基矽氧烷單體)與其餘實施例及比較例(分別使用0.5mol%、2mol%、4.9mol%、7mol%、10mol%及12mol%的二氨基矽氧烷單體(以二胺單體的總摩爾數計))所得聚醯亞胺層的玻璃化轉換溫度,可知添加二氨基矽氧烷單體可降低聚醯亞胺層的玻璃化轉換溫度。
而比較例17至18及比較例B17至B18的結果顯示,使用12mol%的二氨基矽氧烷單體時,不僅玻璃化轉換溫度下降至245℃至251℃、抗張強度不佳,且無法通過UL94V0等級的阻燃測試,阻燃性能不佳。
比較例16與B16的結果顯示,若未添加二氨基矽氧烷單體,兩聚醯亞胺層之間無法有效附著在一起。
最後應說明的是:以上各實施例僅用以說明本發明的技術方案,而非對其限制;儘管參照前述各實施例對本發明進行了詳細的說明,本領域的普通技術人員應當理解:其依然可以對前述各實施例所記載的技術方案進行修改,或者對其中部分或者全部技術特徵進行等同替換;而這些修改或者替換,並不使相應技術方案的本質脫離本發明各實施例技術方案的範圍。