一種帶三氟甲硫基的茚酮類化合物及其製備方法與流程
2024-02-14 07:09:15 3
本發明屬於有機化學合成技術領域,涉及一種帶三氟甲硫基的茚酮類化合物及其製備方法。
背景技術:
三氟甲硫基(-SCF3) 具有強吸電子效應、高親脂性(疏水性參數)和良好的生物活性等特性, 故將其引入到有機化合物中能夠顯著改變化合物化學穩定性和代謝穩定性。因此,含有-SCF3基的化合物在醫藥,農用化學品和材料等領域具有極大的潛在應用價值。例如,抗球蟲藥物妥曲珠利(Toltrazuril)、殺蟲劑 (Vaniliprole)和食慾減退藥物替氟雷司 (Tiflorex) 、抗高血壓藥氯沙坦(Losartan analogue)等等,都含有三氟甲硫基,如下文化學結構式所示:
基於三氟甲硫基的優良特性,人們對三氟甲硫基化反應顯示了廣泛的興趣。例如,研究人員已經研發製備了多種芳基化,烯化,羰基化等帶有三氟甲硫基化的相關化合物。
芳基化反應,如Harris 發現的三氟甲硫醇CF3SH對氟烯烴的加成反應,該反應在X射線或紫外線照射下生成三氟甲硫基自由基然後對氟烯烴進行加成。但該反應存在原料難以保存、反應條件較為苛刻等缺點;如Billard用N-三氟甲硫基苯胺和富電子的吲哚在對甲苯磺酸和溶劑DCM存在下發生親電反應,進行烯基化反應,生成三氟甲硫基取代的吲哚類化合物;但但由於N-三氟甲硫基苯胺原料難以製備,且其只能和富電子的吲哚反應,因此反應局限性較大;羰基化反應,如Munavalli,S在研究中發現,CF3SCl光照下可以與與三甲基矽基烯醇醚發生自由基反應,生成2-三氟甲硫基環己酮,2,2-二三氟甲硫基環己酮和2,6二三氟甲硫基環己酮等,但其生成產物不專一,並且CF3SCl不穩定,毒性大。鑑於上述芳基化、烯化、羰基化等反應存在以下各種局限性:原料毒性大、原料不穩定、反應條件苛刻、必須用紫外燈或者光照才能參與反應、並且底物範圍適應性並不廣泛等,本發明採用將三氟甲硫基這一活性片段嫁接到茚酮類化合物上的方法來製備一系列帶有三氟甲硫基茚酮類合物。
技術實現要素:
為了克服現有技術的不足,本發明提供了一種綠色環保、安全高效的製備一種帶三氟甲硫基的茚酮類化合物的製備方法,該製備方法解決了直接生成茚酮類化合物過程中存在的成本過高和底物適應性差等的技術難題;且該製備方法具有成本低、穩定性高、反應條件溫和、反應時間短、原料廉價易得、產物收率和純度高等諸多優點。
為了實現上述目的,本發明採用的技術方案是:一種帶三氟甲硫基的茚酮類化合物的製備方法,其特徵在於,包括以下步驟:選取炔酮類化合物和三氟甲硫烷醇銀化合物為原料,以過硫酸鹽試劑作為氧化劑,以六甲基磷醯三胺為穩定劑,在反應溶劑中,於反應溫度為80oC下進行反應,反應12h,反應結束後經後處理獲得如下式(II)的茚酮類化合物,其化學反應式如下所示,
(I) (II)
所述式I中的R1為氫、滷素基、烷氧基中的任意一種。
上述反應溫度非限定地可以為40oC、50oC、60oC、70oC、80oC、90oC、100oC或120oC,優選的是80℃;反應時間非限定性地可以為10小時、12小時、14小時、16小時、18小時或24小時,優選的是12h,或者反應時間還可以通過氣質聯用儀或TLC檢測原料的殘留量多少而確定合適的反應時間;上述滷素性能相近,可以選擇-F、-Cl、-Br、-I中的任意一種;而烷氧基則可以選擇CH3O-、C2H5O-、C3H7O-、C4H9O-、C5H11O-、C6H13O-等等飽和烷氧基中的任意一種。
進一步的,所述過硫酸鹽試劑為過硫酸鉀、過硫酸鈉中的任意一種。
進一步的,所述反應溶劑為乙腈、N,N-二甲基甲醯胺(DMF)、乙醇、二甲基亞碸(DMSO)、乙醇、丙酮中的至少任意一種。優選為二甲基亞碸。
進一步的,所述炔酮類化合物和三氟甲硫烷醇銀化合物的摩爾比為1:1.5。非限定性地可以為1:1、1:1.2、1:1.5、1:1.8或1:2.0,優選的為1:1.5。
進一步的,所述炔酮類化合物和六甲基磷醯三胺穩定劑的摩爾比為1:0.1。
進一步的,所述炔酮類化合物與過硫酸鹽的摩爾比為1:1-5。非限定性地可以為1:1、1:2、1:3、1:4或1:5,兩者優選的摩爾比為1:3-5。
進一步的,反應結束後的後處理包括如下步驟,反應完畢後,用旋轉蒸發儀從反應結束後得到的混合物中除去溶劑,殘留物用300-400目矽膠柱層析提純得到目標產物,柱層析過程可以用TLC跟蹤監控而確定合適的洗脫終點。
本發明的另一個目的是提供了一種由上述的製備方法所製得的帶三氟甲硫基的茚酮類化合物,其特徵在於,包含-SCF3官能團,化學結構式如下所示:
R2為氫。其中,R2還可以是烷基、烷氧基、滷素基等。
採用上述方案,本發明的合成方法以炔酮類化合物和三氟甲硫烷醇銀化合物(AgSCF3由於其穩定性高、經濟易得,是提供三氟甲硫基的理想選擇)為原料,使用過硫酸鉀或過硫酸鈉試劑作為氧化劑以及旋轉合適的工藝參數進行合適的組合,通過過硫酸鹽產生自由基使得一價銀被氧化成二價銀,從而產生三氟甲硫基自由基,進一步環化經過β-H消除,從而一步得到了帶三氟甲硫基的茚酮類化合物;該反應體系中,原料完全轉化,只生成目標產物,沒有其他雜質的產生,原子經濟性高、合成體系成本低、反應條件簡單,並且可以直接在空氣中進行,對於操作人員來說,安全性高;反應結束後的後處理過程除了上述方法,還可以採用結晶、柱層析提純、萃取中的任何一種處理手段或多種處理手段的組合。該製備方法具有反應條件溫和、反應時間短、原料廉價易得、產物收率和純度高等諸多優點,為新型三氟甲硫基螺雜環類化合物的製備提供了有效的合成方法,具有良好的研究價值和應用前景。目前使用一步合成茚酮類化合物的方法,其底物適用性較差,比如上述帶缺電子苯環類化合物就不能反應,因此,並不適用於製備將三氟甲硫基直接導入茚酮類化合物中,而且由於溫度越高,三氟甲硫基越容易氧化,故而導致產率較低,本發明加入了六甲基磷醯三胺為三氟甲硫基的穩定劑,配合過硫酸鹽來提高合成的目標產物的產率,因此本發明解決了目前合成帶三氟甲硫基茚酮類化合物方法中的系列技術難題。
下面結合具體實施例對本發明作進一步描述。
附圖說明
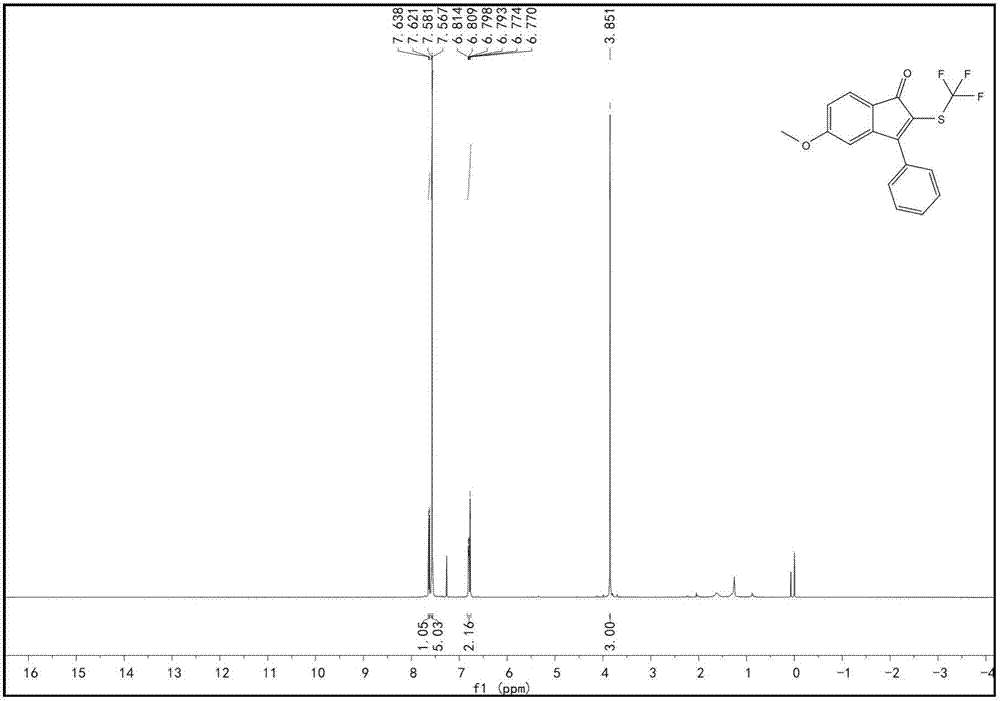
附圖1為本發明具體實施例一至五、實施例十至十三中產物5-甲氧基-3-苯基-2-((三氟甲基)硫)-1H-茚-1-酮核磁譜圖;
附圖2為本發明具體實施例一至五、實施例十至十三中產物5-甲氧基-3-苯基-2-((三氟甲基)硫)-1H-茚-1-酮核磁譜圖;
附圖3為本發明具體實施例一至五、實施例十至十三中產物5-甲氧基-3-苯基-2-((三氟甲基)硫)-1H-茚-1-酮核磁譜圖;
附圖4為本發明具體實施例六中產物7-氯-3-苯基-2-((三氟甲基)硫)-1H-茚-1-酮核磁譜圖;
附圖5為本發明具體實施例六中產物7-氯-3-苯基-2-((三氟甲基)硫)-1H-茚-1-酮核磁譜圖;
附圖6為本發明具體實施例六中產物7-氯-3-苯基-2-((三氟甲基)硫)-1H-茚-1-酮核磁譜圖;
附圖7為本發明具體實施例七中產物3-苯基-2-((三氟甲基)硫)-1H-茚-1-酮核磁譜圖;
附圖8為本發明具體實施例七中產物3-苯基-2-((三氟甲基)硫)-1H-茚-1-酮核磁譜圖;
附圖9為本發明具體實施例七中產物3-苯基-2-((三氟甲基)硫)-1H-茚-1-酮核磁譜圖;
附圖10為本發明具體實施例八中產物5-乙氧基-3-苯基-2-(三氟甲基)硫)-1H-茚-1-酮的核磁譜圖;
附圖11為本發明具體實施例八中產物5-乙氧基-3-苯基-2-(三氟甲基)硫)-1H-茚-1-酮的核磁譜圖;
附圖12為本發明具體實施例八中產物5-乙氧基-3-苯基-2-(三氟甲基)硫)-1H-茚-1-酮的核磁譜圖;
附圖13為本發明具體實施例九中產物5,7-二甲氧基-3-苯-2-(三氟甲基)硫)-1H-茚-1-酮核磁譜圖;
附圖14為本發明具體實施例九中產物5,7-二甲氧基-3-苯-2-(三氟甲基)硫)-1H-茚-1-酮核磁譜圖;
附圖15為本發明具體實施例九中產物5,7-二甲氧基-3-苯-2-(三氟甲基)硫)-1H-茚-1-酮核磁譜圖。
具體實施方式
本發明不局限於上述具體實施方式,本領域一般技術人員根據本發明公開的內容,可以採用其他多種具體實施方式實施本發明的,或者凡是採用本發明的設計結構和思路,做簡單變化或更改的,都落入本發明的保護範圍。
本發明的具體實施例如下所示:
該反應過程如下:選取炔酮類化合物A和三氟甲硫烷醇銀化合物B為原料,以過硫酸鹽試劑作為氧化劑,以六甲基磷醯三胺(HMPA)為穩定劑,在反應溶劑中,於反應溫度為80 oC下進行反應,反應12h,反應結束後經後處理獲得茚酮類化合物。
表1-實施例原料配比
具體實例一:採用1-(4-甲氧基苯基)-3-苯基丙-2-炔-1-酮和三氟甲硫烷醇銀合成5-甲氧基-3-苯基-2-((三氟甲基)硫)-1H-茚-1-酮;
將原料A、原料B、過硫酸鉀,HMPA按摩爾比為1:1.5:1:0.1稱取,其中(A)化合物為0.1 mmol。反應溶液採用DMSO,將反應體系在80oC下攪拌反應12小時。反應結束後冷卻,用飽和氯化鈉水溶液洗進行清洗,無水硫酸鈉除水,短矽膠柱抽濾,濾液旋蒸,除去過硫酸鉀,除去溶劑,剩餘物用矽膠柱層析,石油醚淋洗,TLC檢測,合併含有產物的流出液,旋轉蒸發儀蒸餾除去溶劑,真空乾燥得到棕黃色的目標產物,產率30%,純度為99.1%(HPLC)。核磁共振波譜:1H NMR (500 MHz, CDCl3) δ 7.63 (d, J = 8.0 Hz, 1H), 7.61 – 7.49 (m, 5H), 6.89-6.69 (m, 2H), 3.85 (s, 3H).13C NMR (125 MHz, CDCl3) δ 190.51, 167.94, 164.54 , 145.84, 130.79,130.67,, 128.79, 128.73 (q,J =308,1C,SCF3),128.53, 125.86 , 123.20, 119.42, 112.37, 112.13, 55.93.19F NMR (471 MHz, CDCl3) δ -40.11 (s)。
具體實例二:採用1-(4-甲氧基苯基)-3-苯基丙-2-炔-1-酮和三氟甲硫烷醇銀合成5-甲氧基-3-苯基-2-((三氟甲基)硫)-1H-茚-1-酮;
將原料A、原料B、過硫酸鉀,HMPA按摩爾比為1:1.5:2:0.1稱取,其中(A)化合物為0.1 mmol。反應溶液採用DMSO,將反應體系在80oC下攪拌反應12小時。反應結束後冷卻,用飽和氯化鈉水溶液洗進行清洗,無水硫酸鈉除水,短矽膠柱抽濾,濾液旋蒸,除去過硫酸鉀,除去溶劑,剩餘物用矽膠柱層析,石油醚淋洗,TLC檢測,合併含有產物的流出液,旋轉蒸發儀蒸餾除去溶劑,真空乾燥得到棕黃色的目標產物,產率53%,純度為99.1%(HPLC)。核磁共振波譜:1H NMR (500 MHz, CDCl3) δ 7.63 (d, J = 8.0 Hz, 1H), 7.61 – 7.49 (m, 5H), 6.89-6.69 (m, 2H), 3.85 (s, 3H).13C NMR (125 MHz, CDCl3) δ 190.51, 167.94, 164.54 , 145.84, 130.79,130.67,, 128.79, 128.73 (q,J =308,1C,SCF3),128.53, 125.86 , 123.20, 119.42, 112.37, 112.13, 55.93.19F NMR (471 MHz, CDCl3) δ -40.11 (s) 。
具體實例三:採用1-(4-甲氧基苯基)-3-苯基丙-2-炔-1-酮和三氟甲硫烷醇銀合成5-甲氧基-3-苯基-2-((三氟甲基)硫)-1H-茚-1-酮;
將原料A、原料B、過硫酸鉀,HMPA按摩爾比為1:1.5:3:0.1稱取,其中(A)化合物為0.1 mmol。反應溶液採用DMSO,將反應體系在80oC下攪拌反應12小時。反應結束後冷卻,用飽和氯化鈉水溶液洗進行清洗,無水硫酸鈉除水,短矽膠柱抽濾,濾液旋蒸,除去過硫酸鉀,除去溶劑,剩餘物用矽膠柱層析,石油醚淋洗,TLC檢測,合併含有產物的流出液,旋轉蒸發儀蒸餾除去溶劑,真空乾燥得到棕黃色的目標產物,產率82%,純度為99.1%(HPLC)。核磁共振波譜:1H NMR (500 MHz, CDCl3) δ 7.63 (d, J = 8.0 Hz, 1H), 7.61-7.49 (m, 5H), 6.89-6.69 (m, 2H), 3.85 (s, 3H).13C NMR (125 MHz, CDCl3) δ 190.51, 167.94, 164.54 , 145.84, 130.79,130.67,, 128.79, 128.73 (q,J =308,1C,SCF3),128.53, 125.86 , 123.20, 119.42, 112.37, 112.13, 55.93.19F NMR (471 MHz, CDCl3) δ -40.11 (s) 。
具體實例四:採用1-(4-甲氧基苯基)-3-苯基丙-2-炔-1-酮和三氟甲硫烷醇銀合成5-甲氧基-3-苯基-2-((三氟甲基)硫)-1H-茚-1-酮;
將原料A、原料B、過硫酸鉀,HMPA按摩爾比為1:1.5:4:0.1稱取,其中(A)化合物為0.1 mmol。反應溶液採用DMSO,將反應體系在80oC下攪拌反應12小時。反應結束後冷卻,用飽和氯化鈉水溶液洗進行清洗,無水硫酸鈉除水,短矽膠柱抽濾,濾液旋蒸,除去過硫酸鉀,除去溶劑,剩餘物用矽膠柱層析,石油醚淋洗,TLC檢測,合併含有產物的流出液,旋轉蒸發儀蒸餾除去溶劑,真空乾燥得到棕黃色的目標產物,產率83%,純度為99.1%(HPLC)。核磁共振波譜:1H NMR (500 MHz, CDCl3) δ 7.63 (d, J = 8.0 Hz, 1H), 7.61-7.49 (m, 5H), 6.89-6.69 (m, 2H), 3.85 (s, 3H).13C NMR (125 MHz, CDCl3) δ 190.51, 167.94, 164.54 , 145.84, 130.79,130.67,, 128.79, 128.73 (q,J =308,1C,SCF3),128.53, 125.86 , 123.20, 119.42, 112.37, 112.13, 55.93.19F NMR (471 MHz, CDCl3) δ -40.11 (s)。
具體實例五:採用1-(4-甲氧基苯基)-3-苯基丙-2-炔-1-酮和三氟甲硫烷醇銀合成5-甲氧基-3-苯基-2-((三氟甲基)硫)-1H-茚-1-酮;將原料A、原料B、過硫酸鉀,HMPA按摩爾比為1:1.5:5:0.1稱取,其中(A)化合物為0.1 mmol。反應溶液採用DMSO,將反應體系在80oC下攪拌反應12小時。反應結束後冷卻,用飽和氯化鈉水溶液洗進行清洗,無水硫酸鈉除水,短矽膠柱抽濾,濾液旋蒸,除去過硫酸鉀,除去溶劑,剩餘物用矽膠柱層析,石油醚淋洗,TLC檢測,合併含有產物的流出液,旋轉蒸發儀蒸餾除去溶劑,真空乾燥得到棕黃色的目標產物,產率80%,純度為99.1%(HPLC)。核磁共振波譜:1H NMR (500 MHz, CDCl3) δ 7.63 (d, J = 8.0 Hz, 1H), 7.61-7.49 (m, 5H), 6.89-6.69 (m, 2H), 3.85 (s, 3H).13C NMR (125 MHz, CDCl3) δ 190.51, 167.94, 164.54 , 145.84, 130.79,130.67,, 128.79, 128.73 (q,J =308,1C,SCF3),128.53, 125.86 , 123.20, 119.42, 112.37, 112.13, 55.93.19F NMR (471 MHz, CDCl3) δ -40.11 (s) 。
上述實施例一至五,R1為甲氧基時,即以採用1-(4-甲氧基苯基)-3-苯基丙-2-炔-1-酮和三氟甲硫烷醇銀合成5-甲氧基-3-苯基-2-((三氟甲基)硫)-1H-茚-1-酮;其中變量為炔酮類化合物與過硫酸鹽的摩爾比,其中,當摩爾比為1:3-5時,產率達到80%以上,說明兩者之間的摩爾配比對產率具有重要影響作用。
具體實施例六:R1為氯時,即採用1-(2-氯苯基)-3-苯基丙-2-炔-1-酮和三氟甲硫烷醇銀合成7-氯-3-苯基-2-((三氟甲基)硫)-1H-茚-1-酮;
將原料A、原料B、過硫酸鉀,HMPA按摩爾比為1:1.5:3:0.1稱取,其中(A)化合物為0.1 mmol。反應溶液採用DMSO,將反應體系在80oC下攪拌反應12小時。反應結束後冷卻,用飽和氯化鈉水溶液洗進行清洗,無水硫酸鈉除水,短矽膠柱抽濾,濾液旋蒸,除去過硫酸鉀,除去溶劑,剩餘物用矽膠柱層析,石油醚淋洗,TLC檢測,合併含有產物的流出液,旋轉蒸發儀蒸餾除去溶劑,真空乾燥得到棕黃色的目標產物,產率78%,純度為99.1%(HPLC)。核磁共振波譜:1H NMR (500 MHz, CDCl3) δ 7.56 (d, J = 4.2 Hz, 5H), 7.41-7.31 (m, 2H), 7.15 (d, J = 6.9 Hz, 1H).13C NMR (125 MHz, CDCl3) δ 188.32, 167.30, 144.96, 133.85, 132.40, 131.76, 130.54, 129.80 ,128.38, 128.13(q,J =308,1C,SCF3), 128.03 , 125.56, 121.26, 118.46. 19F NMR (471 MHz, CDCl3) δ -39.83 (s) 。
具體實例七:R1為H時,即採用1,3-二苯-2-炔丙基-1-酮和三氟甲硫烷醇銀合成3-苯基-2-((三氟甲基)硫)-1H-茚-1-酮;
將原料A、原料B、過硫酸鈉,HMPA按摩爾比為1:1.5:3:0.1稱取,其中(A)化合物為0.1 mmol。反應溶液採用DMSO,將反應體系在80oC下攪拌反應12小時。反應結束後冷卻,用飽和氯化鈉水溶液洗進行清洗,無水硫酸鈉除水,短矽膠柱抽濾,濾液旋蒸,除去過硫酸鉀,除去溶劑,剩餘物用矽膠柱層析,石油醚淋洗,TLC檢測,合併含有產物的流出液,旋轉蒸發儀蒸餾除去溶劑,真空乾燥得到棕黃色的目標產物,產率77%,純度為99.1%(HPLC)。核磁共振波譜:1H NMR (500 MHz, CDCl3) δ 7.69-7.65 (m, 1H), 7.62-7.55 (m, 5H), 7.49-7.40 (m, 2H), 7.26-7.22 (m, 1H) 19F NMR (471 MHz, CDCl3) δ -40.24 (s).13C NMR (125 MHz, CDCl3) δ 190.97, 168.96, 142.35, 132.73, 129.94, 129.88, 129.72, 129.70, , 127.77 (s), 128.73 (q,J =308,1C,SCF3), 127.48, 122.79, 122.10, 116.86.19F NMR (471 MHz, CDCl3) δ -40.24 (s) 。
具體實例八:R1為乙氧基時,即採用1-(4-乙氧基)-3-苯基丙-2-炔-1-酮和三氟甲硫烷醇銀合成5-乙氧基-3-苯基-2-(三氟甲基)硫)-1H-茚-1-酮;
將原料A、原料B、過硫酸鉀,HMPA按摩爾比為1:1.5:3:0.1稱取,其中(A)化合物為0.1 mmol。反應溶液採用DMSO,將反應體系在80oC下攪拌反應12小時。反應結束後冷卻,用飽和氯化鈉水溶液洗進行清洗,無水硫酸鈉除水,短矽膠柱抽濾,濾液旋蒸,除去過硫酸鉀,除去溶劑,剩餘物用矽膠柱層析,石油醚淋洗,TLC檢測,合併含有產物的流出液,旋轉蒸發儀蒸餾除去溶劑,真空乾燥得到棕黃色的目標產物,產率85%,純度為99.1%(HPLC)。核磁共振波譜:1H NMR (500 MHz, CDCl3) δ 7.46 (s, 5H), 7.34-7.25 (m, 1H), 6.90 (d, J = 8.6 Hz, 1H), 6.73 (d, J = 7.2 Hz, 1H), 4.20 (q, J = 7.0 Hz, 2H), 1.43 (t, J = 7.0 Hz, 3H).13C NMR (125 MHz, CDCl3) δ 188.37, 166.49, 155.67, 144.57 , 134.53, 129.47, 128.99, 127.75(q,J =308,1C,SCF3),127.62, 117.52, 116.31, 115.29, 114.68, 63.98, 13.70. 19F NMR (471 MHz, CDCl3) δ -40.06 (s) 。
上述實施例三、六至八,原料A、原料B、過硫酸鉀,HMPA之間的配比均按摩爾比為1:1.5:3:0.1,而原料A中的R1分別採用了氫、滷素基、烷氧基,且其產率較為穩定,說明了本發明的底物具有良好的適應性。
具體實例九:1-(2,4-二甲氧基苯)-3-苯丙-2-炔-1-酮和三氟甲硫烷醇銀合成5,7-二甲氧基-3-苯-2-(三氟甲基)硫)-1H-茚-1-酮;
將原料A、原料B、過硫酸鉀,HMPA按摩爾比為1:1.5:3:0.1稱取,其中(A)化合物為0.1 mmol。反應溶液採用DMSO,將反應體系在80oC下攪拌反應12小時。反應結束後冷卻,用飽和氯化鈉水溶液洗進行清洗,無水硫酸鈉除水,短矽膠柱抽濾,濾液旋蒸,除去過硫酸鉀,除去溶劑,剩餘物用矽膠柱層析,石油醚淋洗,TLC檢測,合併含有產物的流出液,旋轉蒸發儀蒸餾除去溶劑,真空乾燥得到棕黃色的目標產物,產率86%,純度為99.1%(HPLC)。核磁共振波譜:1H NMR (500 MHz, CDCl3) δ 7.58 – 7.48 (m, 5H), 6.38 (dd, J = 15.0, 1.8 Hz, 2H), 3.99 (s, 3H), 3.85 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 188.01, 166.39, 165.65, 159.13, 147.44, 130.87, 130.38, 128.73(q,J =308,1C,SCF3),128.66,120.07, 108.87, 105.50, 98.58, 56.32, 55.98. 19F NMR (471 MHz, CDCl3) δ -39.99 (s) 。
具體實例十:R1為甲氧基時,即採用1-(4-甲氧基苯基)-3-苯基丙-2-炔-1-酮和三氟甲硫烷醇銀合成5-甲氧基-3-苯基-2-((三氟甲基)硫)-1H-茚-1-酮;
將原料A、原料B、過硫酸鉀,HMPA按摩爾比為1:1.5:3:0.1稱取,其中(A)化合物為0.1 mmol。反應溶液採用乙醇,將反應體系在80oC下攪拌反應12小時。反應結束後冷卻,用飽和氯化鈉水溶液洗進行清洗,無水硫酸鈉除水,短矽膠柱抽濾,濾液旋蒸,除去過硫酸鉀,除去溶劑,剩餘物用矽膠柱層析,石油醚淋洗,TLC檢測,合併含有產物的流出液,旋轉蒸發儀蒸餾除去溶劑,真空乾燥得到棕黃色的目標產物,產率30%,純度為99.1%(HPLC)。核磁共振波譜:1H NMR (500 MHz, CDCl3) δ 7.63 (d, J = 8.0 Hz, 1H), 7.61 – 7.49 (m, 5H), 6.89 – 6.69 (m, 2H), 3.85 (s, 3H).13C NMR (125 MHz, CDCl3) δ 190.51, 167.94, 164.54 , 145.84, 130.79,130.67,, 128.79, 128.73 (q,J =308,1C,SCF3),128.53, 125.86 , 123.20, 119.42, 112.37, 112.13, 55.93.19F NMR (471 MHz, CDCl3) δ -40.11 (s) 。
具體實例十一:R1為甲氧基時,採用1-(4-甲氧基苯基)-3-苯基丙-2-炔-1-酮和三氟甲硫烷醇銀合成5-甲氧基-3-苯基-2-((三氟甲基)硫)-1H-茚-1-酮;
將原料A、原料B、過硫酸鉀,HMPA按摩爾比為1:1.5:3:0.1稱取,其中(A)化合物為0.1 mmol。反應溶液採用CH3CN,將反應體系在80oC下攪拌反應12小時。反應結束後冷卻,用飽和氯化鈉水溶液洗進行清洗,無水硫酸鈉除水,短矽膠柱抽濾,濾液旋蒸,除去過硫酸鉀,除去溶劑,剩餘物用矽膠柱層析,石油醚淋洗,TLC檢測,合併含有產物的流出液,旋轉蒸發儀蒸餾除去溶劑,真空乾燥得到棕黃色的目標產物,產率57%,純度為99.1%(HPLC)。核磁共振波譜:1H NMR (500 MHz, CDCl3) δ 7.63 (d, J = 8.0 Hz, 1H), 7.61 – 7.49 (m, 5H), 6.89 – 6.69 (m, 2H), 3.85 (s, 3H).13C NMR (125 MHz, CDCl3) δ 190.51, 167.94, 164.54 , 145.84, 130.79,130.67,, 128.79, 128.73 (q,J =308,1C,SCF3),128.53, 125.86 , 123.20, 119.42, 112.37, 112.13, 55.93.19F NMR (471 MHz, CDCl3) δ -40.11 (s) 。
具體實例十二:採用1-(4-甲氧基苯基)-3-苯基丙-2-炔-1-酮和三氟甲硫烷醇銀合成5-甲氧基-3-苯基-2-((三氟甲基)硫)-1H-茚-1-酮;
將原料A、原料B、過硫酸鉀,HMPA按摩爾比為1:1.5:3:0.1稱取,其中(A)化合物為0.1 mmol。反應溶液採用DMF,將反應體系在80oC下攪拌反應12小時。反應結束後冷卻,用飽和氯化鈉水溶液洗進行清洗,無水硫酸鈉除水,短矽膠柱抽濾,濾液旋蒸,除去過硫酸鉀,除去溶劑,剩餘物用矽膠柱層析,石油醚淋洗,TLC檢測,合併含有產物的流出液,旋轉蒸發儀蒸餾除去溶劑,真空乾燥得到棕黃色的目標產物,產率45%,純度為99.1%(HPLC)。核磁共振波譜:1H NMR (500 MHz, CDCl3) δ 7.63 (d, J = 8.0 Hz, 1H), 7.61 – 7.49 (m, 5H), 6.89 – 6.69 (m, 2H), 3.85 (s, 3H).13C NMR (125 MHz, CDCl3) δ 190.51, 167.94, 164.54 , 145.84, 130.79,130.67,, 128.79, 128.73 (q,J =308,1C,SCF3),128.53, 125.86 , 123.20, 119.42, 112.37, 112.13, 55.93.19F NMR (471 MHz, CDCl3) δ -40.11 (s) 。
具體實例十三:採用1-(4-甲氧基苯基)-3-苯基丙-2-炔-1-酮和三氟甲硫烷醇銀合成5-甲氧基-3-苯基-2-((三氟甲基)硫)-1H-茚-1-酮;
將原料A、原料B、過硫酸鉀,HMPA按摩爾比為1:1.5:3:0.1稱取,其中(A)化合物為0.1 mmol。反應溶液採用丙酮,將反應體系在80oC下攪拌反應12小時。反應結束後冷卻,用飽和氯化鈉水溶液洗進行清洗,無水硫酸鈉除水,短矽膠柱抽濾,濾液旋蒸,除去過硫酸鉀,除去溶劑,剩餘物用矽膠柱層析,石油醚淋洗,TLC檢測,合併含有產物的流出液,旋轉蒸發儀蒸餾除去溶劑,真空乾燥得到棕黃色的目標產物,產率15%,純度為99.1%(HPLC)。核磁共振波譜:1H NMR (500 MHz, CDCl3) δ 7.63 (d, J = 8.0 Hz, 1H), 7.61 – 7.49 (m, 5H), 6.89 – 6.69 (m, 2H), 3.85 (s, 3H).13C NMR (125 MHz, CDCl3) δ 190.51, 167.94, 164.54 , 145.84, 130.79,130.67,, 128.79, 128.73 (q,J =308,1C,SCF3),128.53, 125.86 , 123.20, 119.42, 112.37, 112.13, 55.93.19F NMR (471 MHz, CDCl3) δ -40.11 (s) 。
上述實施例十-十三與實施例三對比可知,原料A、原料B、過硫酸鉀,HMPA之間的配比均按摩爾比為1:1.5:3:0.1,而原料A中的R1均採用甲氧基,其變量則是選取不同的反應溶劑,由上述實施例可知,溶劑的選擇對產率具有重大影響,即使一些性能相似的溶劑,如DMF與DMSO,其對本發明的產率具有不同的結果,故本發明的溶劑選擇是基於技術人員創造性的實驗基礎上得出的,不具有普遍性,只具有特殊適應性。