改性無機耐熱氧化物及其製備方法及加氫脫硫催化劑和降低加氫脫硫催化劑失活速度的方法與流程
2023-11-11 11:58:33 1
本發明是關於一種改性無機耐熱氧化物及其製備方法和應用以及使用該改性無機耐熱氧化物作為載體的加氫脫硫催化劑和降低加氫脫硫催化劑失活速度的方法。
背景技術:
加氫處理是現代煉油工業中的支柱技術,在生產清潔燃料、提高產品質量、充分利用石油資源和原料預處理等方面發揮著重要作用。隨著經濟、環保和社會的發展,使得煉油企業對加氫處理催化劑的活性和穩定性不斷提出更高的要求。常見加氫處理催化劑以VIII族金屬Co、Ni作助活性組元,以VIB族金屬Mo、W作主活性組元,載體常選用氧化鋁。
例如,CN201010236483.7公開了一種以氧化矽-氧化鋁為載體的加氫脫硫催化劑,含有氧化鈷2~6wt%,氧化鉬9~15wt%,鹼土金屬氧化物2~8wt%,氧化磷2~6wt%,鹼金屬氧化物3~5wt%,氧化矽2~6wt%,氧化鋁54~80wt%。催化劑的加氫活性和選擇性高,穩定性好。
CN201010269179.2描述了一種加氫脫硫催化劑及其製備方法,所述催化劑是以鉬、鎳、鋅、銅複合金屬氧化物至少兩種為活性組分,添加Si、Ti、Ca、Ce、Mg、P氧化物至少一種為助劑,餘量為氧化鋁。
CN200710177578.4涉及一種組合氧化鋁基的選擇性加氫脫硫催化劑及其製備方法,該方法將大孔氧化鋁和小孔氧化鋁複合,然後依次負載助劑鎂和硼以及活性組分鈷和鉬,再經乾燥和焙燒,製成催化劑。
CN200710177577.X公開了一種催化劑以氧化鋁和氧化硼為載體,並經 過多元助劑鎂、鉀和磷修飾,擔載活性組分鈷和鉬。
CN200480005056.0公開了一種含有非貴金屬的第VIII族金屬、一種非貴金屬的第VIB族金屬和一或多種選自鋁、矽、鎂、鈦、鋯、硼和鋅的元素的催化劑組合物。
CN201210268290.9公開了載體由氧化鋁、氧化矽、氧化鎂組成、而所述活性組分由WO3和NiO或MoO3和NiO組成、並含有以K2O和Ga2O3組成的助劑的催化劑。
CN96120875.9公開了含有活性組分Ni、W、Co,助催化劑成分為P或P和Mg的混合物,載體為γ-Al2O3。
CN96106586.9公開了一種催化劑,它由γ-Al2O3負載Ni、W、Co活性組分,還可以加入選自Mg、Zn、Fe、Ca中的任一元素作為助催化劑成分。
儘管現有技術對催化劑的組成及其製備方法進行了大量的研究,催化劑的性能也取得了較大的進步。然而,隨著社會對燃料清潔程度要求的增加以及原油劣質化加劇的趨勢,加氫處理催化劑的性能需要進一步提高。
技術實現要素:
為了克服現有技術的不足,本發明提供了一種能夠製備性能更為優越的催化劑的改性無機耐熱氧化物及其製備方法和應用及加氫脫硫催化劑。
文獻報導,在氧化鋁的表面存在多種具有不同強度的酸性羥基和鹼性羥基(C.Morterra et al.,Catalysis Today 27(1996)497-532)。在氧化態催化劑中,主活性組分(如Mo或W)以氧化物的形式分散在載體的表面,Mo或W物種與氧化鋁表面的各種羥基作用形成化學鍵。形成的化學鍵在硫化過程中發生斷裂並釋放出金屬,釋放的金屬與硫發生反應生成硫化態活性相。硫化態活性相的分散性質對催化劑的活性具有重要影響。本發明的發明人發現,通過優化載體表面羥基的性質,能夠使活性組分更好的分散,從而獲得更多活 性中心,由此在實際應用過程中催化劑活性更高,並且催化劑的失活速率得到降低,可以延長催化劑的使用壽命。
基於此,根據本發明的第一方面,本發明提供了一種改性無機耐熱氧化物,該改性無機耐熱氧化物具有酸性羥基和鹼性羥基,紅外光譜中,強酸性羥基峰在3640-3700cm-1附近,強鹼性羥基峰在3740-3800cm-1附近,其特徵在於,所述強鹼性羥基峰的相對高度為30-90%,所述強鹼性羥基峰與所述強酸性羥基峰的峰高比為1:0.85-1.15。
根據本發明的第二方面,本發明提供了一種改性無機耐熱氧化物的製備方法,該方法包括將無機耐熱氧化物和/或無機耐熱氧化物的前身物、助擠劑、膠溶劑混合均勻後進行成型,然後將所得成型物依次進行乾燥和焙燒,其特徵在於,該方法還包括在成型前和/或成型後加入酸性羥基中和劑和鹼性羥基中和劑,所述酸性羥基中和劑為鹼土金屬氧化物、鹼土金屬硝酸鹽和鹼土金屬氫氧化物中的一種或多種,所述鹼性羥基中和劑為含硼、鎵、矽、鍺、磷、氟元素中的一種或多種的氧化物、含氧酸中的一種或多種,所述酸性羥基中和劑和鹼性羥基中和劑的用量使得所得耐熱無機氧化物的紅外光譜中,強鹼性羥基峰的相對高度為30-90%,強酸性羥基峰的相對高度為20-90%,所述強酸性羥基峰在3640-3700cm-1附近,強鹼性羥基峰在3740-3800cm-1附近。
根據本發明的第三方面,本發明提供了一種由上述製備方法製得的改性無機耐熱氧化物。
根據本發明的第四方面,本發明提供了一種由上述改性無機耐熱氧化物作為催化劑載體的應用。
根據本發明的第五方面,本發明提供了一種加氫脫硫催化劑,該加氫脫硫催化劑含有載體和負載在載體上的金屬活性成分,其特徵在於,所述載體為本發明上述提供的改性無機耐熱氧化物。
根據本發明的第六方面,本發明提供了一種降低加氫脫硫催化劑失活速 度的方法,該方法包括採用上述方法製備改性無機耐熱氧化物,然後以該改性無機耐熱氧化物為載體負載具有加氫脫硫活性的金屬活性成分。
本發明提供的改性無機耐熱氧化物由於強鹼性羥基峰與強酸性羥基峰的峰高相當,使得其作為載體時的催化劑活性更高,並且催化劑的失活速率得到降低,可以延長催化劑的使用壽命。本發明提供的加氫脫硫催化劑通過使用上述改性無機耐熱氧化物作為載體並進一步配合特定的金屬活性組分含量,從而催化劑的活性進一步明顯提高,催化劑的失活速率進一步明顯降低。
本發明的其它特徵和優點將在隨後的具體實施方式部分予以詳細說明。
附圖說明
附圖是用來提供對本發明的進一步理解,並且構成說明書的一部分,與下面的具體實施方式一起用於解釋本發明,但並不構成對本發明的限制。在附圖中:
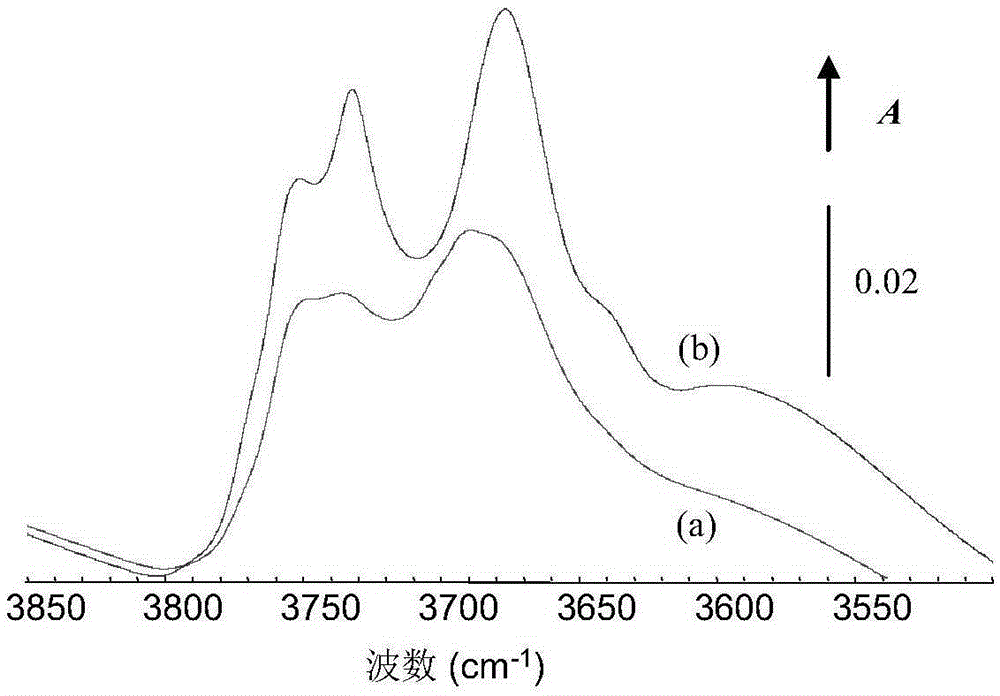
圖1中(a)是實施例1製得的氧化鋁的紅外光譜曲線,(b)是對比例1製得的氧化鋁的紅外光譜圖。
具體實施方式
以下對本發明的具體實施方式進行詳細說明。應當理解的是,此處所描述的具體實施方式僅用於說明和解釋本發明,並不用於限制本發明。
根據本發明的第一方面,本發明提供了一種改性無機耐熱氧化物,該改性無機耐熱氧化物具有酸性羥基和鹼性羥基,紅外光譜中,強酸性羥基峰在3640-3700cm-1附近,強鹼性羥基峰在3740-3800cm-1附近,其特徵在於,所述強鹼性羥基峰與所述強酸性羥基峰的峰高比為1:0.85-1.15,優選為1:0.9-1.1。
本發明中,所述強鹼性羥基峰與所述強酸性羥基峰的峰高相當,由此所述強鹼性羥基與所述強酸性羥基數量相當,使得其作為載體時的催化劑活性更高,並且催化劑的失活速率得到降低,可以延長催化劑的使用壽命。而現有技術中,無機耐熱氧化物的所述強鹼性羥基峰與所述強酸性羥基峰的峰高比通常為1:1.2-1.4。
優選情況下,本發明提供的無機耐熱氧化物的紅外譜圖中,所述強鹼性羥基峰的相對高度為30-90%,優選為40-85%,更優選為55-75%;所述強酸性羥基峰的相對高度為20-90%,優選為50-85%,更優選為60-80%。
本發明中,羥基峰的相對高度的含義與現有相對結晶度的含義類似,具體表示本發明的改性無機耐熱氧化物的羥基峰的高度與參比無機耐熱氧化物的相應羥基峰的高度比,即,相對於參比無機耐熱氧化物,鹼性羥基峰高度降低幅度控制為10-70%,優選為15-60%;強酸性羥基峰的降低幅度控制為10-80%,優選為15-40%。所謂參比無機耐熱氧化物是指製備過程中不引入鹼性羥基中和劑和酸性羥基中和劑時製備的無機耐熱氧化物,即常規的無機耐熱氧化物。
本發明中,兩種酸性羥基變化幅度可以相同,也可以不同,只要最終的高度比或相對高度控制在上述範圍內即可。由於氧化鋁表面中還存在其他類型羥基,如弱酸性和弱鹼性羥基,在調變過程中這些羥基的數量可能發生變化,其變化量不作為控制指標。另外新的羥基也可能生產,屬於正常現象,不做限制。
本發明中,所述紅外光譜圖是首先採用自支撐壓片法獲得樣品,然後在紅外池中對樣品進行預處理後再用紅外光譜儀掃描得到樣品的羥基譜圖(以下稱為自支撐壓片法)。自支撐壓片法為本領域熟知技術,不再贅述。
在進行載體羥基測量前,對樣品進行預處理,預處理在400℃下進行3h以上,處理後將溫度降至室溫。該測量方法為本領域中常用技術,在此不再 詳述。
根據本發明提供的改性無機耐熱氧化物可以是現有技術各種能夠用作催化劑載體的無機耐熱氧化物的改性物,例如,可以為氧化鋁、氧化矽、氧化鋯、氧化鋁-氧化鈦和氧化鋁-氧化矽中的一種或多種的改性物。所謂的改性物即為強鹼性羥基與弱鹼性羥基數量相當從而強鹼性羥基峰與所述強酸性羥基峰的峰高比為1:0.85-1.15,優選為1:0.9-1.1並且進一步優選鹼性羥基和酸性羥基均有所弱化的產物。
所述無機耐熱氧化物改性前後的形狀均可以是現有技術已知的各種使用用作催化劑載體的形狀,例如可以為顆粒狀、條狀或三葉草狀,且無機耐熱氧化物的大小為0.2-15毫米。
本發明中,(改性)無機耐熱氧化物的大小是指無機耐熱氧化物從一點到另一點的最大直線距離,可以通過篩分獲得。
根據本發明的第二方面,本發明提供了一種無機耐熱氧化物的製備方法,該方法包括將無機耐熱氧化物和/或無機耐熱氧化物的前身物、助擠劑、膠溶劑混合均勻後進行成型,然後將所得成型物依次進行乾燥和焙燒,其特徵在於,該方法還包括在成型前和/或成型後加入酸性羥基中和劑和鹼性羥基中和劑,所述酸性羥基中和劑為鹼土金屬氧化物、鹼土金屬硝酸鹽和鹼土金屬氫氧化物中的一種或多種,所述鹼性羥基中和劑為含硼、鎵、矽、鍺、磷、氟元素中的一種或多種的氧化物、含氧酸中的一種或多種,所述酸性羥基中和劑和鹼性羥基中和劑的用量使得所得耐熱無機氧化物的強鹼性羥基峰的相對高度為30-90%,強酸性羥基峰的相對高度為20-90%,所述強酸性羥基峰在3640-3700cm-1附近,強鹼性羥基峰在3740-3800cm-1附近。
需要說明的是,本發明的中和並非通常意義上的酸鹼中和,而是改變羥基性質的中和,具體的是降低羥基峰高度的反應。例如,儘管硝酸具有酸性,能夠中和鹼性物質,但並不能用作本發明的鹼性羥基中和劑。實驗證明,無 論是加大作為膠溶劑時的硝酸的用量,還是在後續混合後或成型過程中額外加入硝酸,均不能起到降低羥基峰高度的作用。
根據本發明,優選情況下,所述酸性羥基中和劑為氫氧化鈹、氫氧化鎂、氫氧化鈣、氫氧化鋇、氧化鈹、氧化鎂、氧化鈣、氧化鍶、氧化鋇、硝酸鋇、硝酸鈣、硝酸鎂、硝酸鈹中的一種或多種,進一步優選為氫氧化鈹、硝酸鈣和硝酸鎂中的一種或多種。
根據本發明,優選情況下,所述鹼性羥基中和劑為HF、氟矽酸、矽溶膠、硼酸、磷酸、亞磷酸、鍺酸中的一種或多種,進一步優選為HF、氟矽酸、矽溶膠、硼酸中的一種或多種。
根據本發明提供的改性無機耐熱氧化物的製備方法,相對於1摩爾的無機耐熱氧化物的前身物,所述酸性羥基中和劑的用量為0.02-0.08摩爾,所述鹼性羥基中和劑的用量為0.02-0.1摩爾。本發明的發明人發現,通過將中和劑的用量控制在上述範圍內,基本可以使鹼性羥基下降幅度在前述值範圍內(30-90%),使酸性羥基下降幅度在前述值範圍內(20-90%)。由於不同的無機耐熱氧化物中羥基量不同,可以通過進一步實驗確定使所述強鹼性羥基峰與所述強酸性羥基峰的峰高比為1:0.85-1.15,優選為1:0.9-1.1範圍內的具體的用量。具體可以參照以下方法來確定:
按照常規方法製備純氧化鋁載體,然後採用孔飽和浸漬法或混捏法製備添加元素的摩爾分數為5%的載體,利用紅外光譜法測量強鹼性或強酸性羥基的相對高度。利用兩點直線法確定相對高度在所需範圍內時元素的用量。例如,氧化鋁中引入摩爾分數為5%的硼,對應強鹼性羥基相對高度為53%。由於強鹼性羥基相對高度與元素含量成反比例關係,因此可以計算出強鹼性羥基相對高度為30-90%時對應的硼元素的摩爾分數。同理,其它元素相應的羥基相對高度範圍的摩爾分數範圍也可以實際測出。
需要特別強調的是,必須嚴格控制羥基中和劑的用量大於上述範圍的下 限值且小於上述範圍的上限值,才能獲得降低羥基峰高度且降低幅度在上述範圍內的效果。事實上,現有技術也在載體的製備過程中加入B、P和Mg作為助劑成分,但它們或者只加入其中同一種性質如僅加入酸性中和劑或者僅加入鹼性中和劑;或者加入的量較大或者較小,因而不能達到同時降低酸性羥基和鹼性羥基峰高度的效果,因此催化劑的加氫脫硫性能也就不能獲得明顯提升。
根據本發明提供的改性無機耐熱氧化物的製備方法,所述酸性羥基中和劑和鹼性羥基中和劑可以同時加入,也可以各自單獨且不同時加入,可以先加酸性羥基中和劑,後加鹼性羥基中和劑,也可以先加鹼性羥基中和劑,後加酸性羥基中和劑。它們可以在製備過程中的各個步驟加入,例如,可以與無機耐熱氧化物的前身物、助擠劑和膠溶劑一起混合均勻後進行成型;也可以在無機耐熱氧化物的前身物、助擠劑和膠溶劑一起混合均勻後再先後加入,然後進行成型;還可以在無機耐熱氧化物的前身物、助擠劑和膠溶劑一起混合均勻後加入酸性羥基中和劑或鹼性羥基中和劑,成型、乾燥並焙燒後再加入鹼性羥基中和劑或酸性羥基中和劑。成型後再加入的方式優選為孔飽和浸漬法。如果在焙燒成型後再加入鹼性羥基中和劑或酸性羥基中和劑,則需要對載體再次進行焙燒。並且第二次焙燒的溫度要低於前一次焙燒溫度,例如低50-200℃,其餘條件保持相同。
本發明的製備方法中,成型的方式可以採用現有的各種催化劑載體成型方法,例如可以是擠條成型法。
成型後可以直接焙燒,也可以先乾燥後焙燒,優選為先乾燥後焙燒。乾燥的溫度可以為60-150℃,優選為100-140℃;乾燥的時間可以為1-10小時,優選為3-6小時。焙燒的溫度可以為300-800℃,優選為400-700℃;焙燒的時間可以為2-10小時,優選為3-5小時。
根據本發明提供的改性無機耐熱氧化物的製備方法,所述無機耐熱氧化 物和/或其前身物可以根據所需的改性無機耐熱氧化物進行適當的選擇,具體的,所述無機耐熱氧化物和/或其前身物可以為擬薄水鋁石、薄水鋁石、無定型氫氧化鋁、三水鋁石、氧化矽、氧化鋯、氧化鋁-氧化鈦和氧化鋁-氧化矽中的一種或多種。
所述助擠劑和膠溶劑均可以參照現有技術進行選擇,例如,助擠劑可以為田菁粉、甲基纖維素和澱粉等物質中的一種或多種,膠溶劑可以為硝酸、檸檬酸和醋酸中的一種或多種。
本發明第三方面還提供了由上述製備方法製得的改性無機耐熱氧化物。
本發明第四方面還提供了改性無機耐熱氧化物作為催化劑載體的應用。
採用本發明提供的上述改性無機耐熱氧化物作為催化劑載體,能夠有效提高催化劑的加氫脫硫活性。
為此,本發明第五方面還提供了一種加氫脫硫催化劑,該加氫脫硫催化劑含有載體和負載在載體上的金屬活性成分,其特徵在於,所述載體為本發明上述提供的改性無機耐熱氧化物。
本發明第六方面還提供了一種降低加氫脫硫催化劑失活速度的方法,該方法包括採用上述方法製備改性無機耐熱氧化物,然後以該改性無機耐熱氧化物為載體負載具有加氫脫硫活性的金屬活性成分。
所述加氫脫硫催化劑的金屬活性成分可以為各種具有加氫脫硫活性的金屬組分,例如可以為元素周期表中VIII族和VIB族金屬元素,優選地,所述金屬活性成分為鎳和/或鈷以及鎢和/或鉬。
所述加氫脫硫催化劑中,載體表面VIB族金屬元素的原子濃度為2-8個原子/nm2,優選為2.5-6個原子/nm2;VIII/(VIII+VIB)原子比範圍是0.2-0.4,優選為0.3-0.4。當所述金屬活性成分為鎳和/或鈷以及鎢和/或鉬時,載體表面Mo和/或W的總原子濃度為2-8個原子/nm2,優選為2.5-6個原子/nm2;Co(Ni)/(Co(Ni)+Mo+W)原子比範圍是0.2-0.4,優選為0.3-0.4。
本發明中,載體表面金屬原子濃度由金屬的原子總數與載體比表面積之比計算而得,載體的比表面積由常規氮氣吸附法測得。
上述加氫脫硫催化劑可以通過將加氫活性金屬組分負載在上述改性無機耐熱氧化物上來製得。例如可以通過孔飽和浸漬法。具體可以通過下述兩種方式來負載。方式1:以硝酸鈷、硝酸鎳、七鉬酸銨和偏鎢酸銨為原料,在添加或者不添加助溶劑的情況下配製成浸漬溶液。助溶劑主要包括氨水,含氮有機化合物,如乙二胺、EDTA、氨基三乙酸、環己二胺四乙酸和胺基酸類。這些物質與Co(Ni)的摩爾比為0.5-2。方式2:以鹼式碳酸鈷、鹼式碳酸鎳、氧化鉬、偏鎢酸銨和磷酸為原料,或添加一種和/或多種含有羥基和/或羧基的有機化合物為助溶劑配製浸漬液。優選選用酒石酸、檸檬酸、乙二醇、丙三醇等。磷與Co(Ni)的原子比為0.5-2,有機物與Co(Ni)的原子比為0.5-2。以上述方法配製的浸漬液浸漬載體0.5-4h之後,在50-180℃下乾燥1-5h,然後在300-500℃下焙燒2-5h製備成催化劑。
本發明提供的加氫脫硫催化劑由於其中的強鹼性和強酸性羥基數量降低,一方面使得活性組分Mo或W與羥基的作用力降低提高了其可硫化能力,另一方面羥基數量的減少使得結焦物質在催化劑上的吸附性能降低進而降低了催化劑的失活速率,可以延長運轉周期。同時,由於強鹼性和強酸性羥基數量的減少,使得載體表面的羥基酸鹼性趨向更加均勻,進而使活性組分更為均勻的分散。由此方法製備催化劑的加氫脫硫活性以及活性穩定性更為優越。
下面的實施例將對本發明做進一步的說明,但是這些實施例並不能限制本發明。以下實施例中使用的氫氧化鋁粉為長嶺催化劑廠生產的擬薄水鋁石。紅外光譜圖是採用自支撐壓片法得到。載體比表面積採用氮氣吸附法測得,金屬原子濃度則根據載體比表面積來確定。
對比例1
將擬薄水鋁石(長嶺催化劑廠生產的PB90粉,比表面積為345m2/g)、田菁粉按照100g:3g比例混合均勻後,加入105mL硝酸(1.5%)溶液,並再次攪拌均勻,然後將其擠條成型。經過在120℃下乾燥3h和在600℃下焙燒4h步驟,製備得到粒徑為1.6mm的純氧化鋁載體D1。載體D1的比表面積為283m2/g。
實施例1
將擬薄水鋁石(同對比例1)、田菁粉按照100g:3g比例混合,並加入1.02g的氫氧化鈹,混合均勻,加入105mL含硝酸(濃度為1.5重量%)和0.47g的HF的水溶液,並再次攪拌均勻,然後將其擠條成型。經過在120℃下乾燥3h和在600℃下焙燒4h步驟,製備得到粒徑為1.6mm的氧化鋁載體Sup1。載體Sup1的比表面積為275m2/g。
利用紅外光譜法考察上述對比例1和實施例1獲得的載體的羥基振動特徵,譜圖如圖1所示。從圖1可以看出,Al2O3載體的強鹼性羥基和強酸性羥基振動波數分別在3780-3750cm-1和3700-3650cm-1。相比於純氧化鋁載體D1,Sup1載體中強鹼性羥基和強酸性羥基的相對高度分別為40%和35%,強鹼性羥基和強酸性羥基的峰高比為1:1.14;而純氧化鋁載體D1中,強鹼性羥基和強酸性羥基的峰高比為1:1.3。
對比例2
將擬薄水鋁石(長嶺催化劑廠生產的PB100粉,比表面積為330m2/g)、田菁粉按照100g:3.5g比例混合均勻後,加入120mL含硝酸(濃度為1.5重量%)的溶液,並再次攪拌均勻擠條成型。經過在120℃下乾燥3h和在550℃下焙燒4h步驟,製備得到粒徑為1.6mm的純氧化鋁載體D2。載體D2 的比表面積為292m2/g。
實施例2
將擬薄水鋁石(同對比例2)、田菁粉按照100g:3.5g比例混合均勻後,加入120mL含硝酸(濃度為1.5重量%)和硼酸(1.21g)的水溶液,並再次攪拌均勻擠條成型。載體經過在120℃下乾燥3h和在600℃下焙燒4h後得到氧化鋁載體,焙燒後的載體以孔飽和浸漬法引入9.01g的硝酸鈣,然後經過120℃乾燥3h和550℃焙燒4h,得到Sup2載體。載體Sup2的比表面積為284m2/g。
利用紅外光譜法考察上述載體的羥基振動特徵發現,相比於純氧化鋁載體D2,Sup2中載體的強鹼性羥基和強酸性羥基的相對高度分別為80%和55%,強鹼性羥基和強酸性羥基的峰高比為1:0.96;而純氧化鋁載體D2中,強鹼性羥基和強酸性羥基的峰高比為1:1.4。
對比例3
將擬薄水鋁石(長嶺催化劑廠生產的PB110粉,比表面積為325m2/g)、田菁粉按照100g:2.5g比例混合均勻後,加入120mL含硝酸(濃度為2重量%)的溶液,並再次攪拌均勻擠條成型。經過在100℃下乾燥5h和在650℃下焙燒3h步驟,製備得到粒徑為1.6mm的純氧化鋁載體D3。載體D3的比表面積為264m2/g。
實施例3
將擬薄水鋁石(同對比例3)、田菁粉按照100g:2.5g比例混合均勻後,首先加入矽溶膠(含7.07g的SiO2)攪拌均勻,而後加入120mL的稀硝酸(濃度為2重量%),並再次攪拌均勻並擠條成型。載體經過在100℃下乾燥5h 和在650℃下焙燒3h後得到氧化鋁載體,焙燒後的載體以孔飽和浸漬法引入6.98g的硝酸鎂,然後經過120℃乾燥3h和550℃焙燒4h,得到Sup3載體。載體Sup3的比表面積為253m2/g。
利用紅外光譜法考察上述對比例3和實施例3獲得的載體的羥基振動特徵發現,Sup3中載體的強鹼性羥基和強酸性羥基的相對高度分別為70%和60%,且強鹼性羥基和強酸性羥基的峰高比為1:1.07;而純氧化鋁載體D3中,強鹼性羥基和強酸性羥基的峰高比為1:1.25。
實施例4
與實施例3相似,將擬薄水鋁石(同實施例3)、田菁粉按照100g:2.5g比例混合均勻後,首先加入矽溶膠(含18.9g的SiO2)攪拌均勻,而後加入110mL的稀硝酸(濃度為2重量%),並再次攪拌均勻並擠條成型。載體經過在100℃下乾燥5h和在650℃下焙燒3h後得到氧化鋁載體,焙燒後的載體以孔飽和浸漬法引入11.64g的硝酸鎂,然後經過120℃乾燥3h和550℃焙燒4h,得到Sup4載體。載體Sup4的比表面積為248m2/g。
利用紅外光譜法考察測得Sup4中載體的強鹼性羥基和強酸性羥基的相對高度分別為30%和21%,且強鹼性羥基和強酸性羥基的峰高比為1:0.88。
對比例4
按照實施例3的方法製備氧化鋁載體,不同的是,硝酸鎂的用量降低至1.75g量,得到對比載體D4。載體D4的比表面積為260m2/g。
利用紅外光譜法考察上述對比例3和對比例4獲得的載體的羥基振動特徵發現,D4中載體的強鹼性羥基和強酸性羥基相對高度分別為85%和95%,且強鹼性羥基和強酸性羥基的峰高比為1:1.39。
對比例5
按照實施例1的方法製備氧化鋁載體,不同的是,HF酸由相同重量的硝酸代替,得到對比載體D5。載體D5的比表面積為261m2/g。
利用紅外光譜法考察上述對比例1和對比例5獲得的載體的羥基振動特徵發現,D5中載體的強鹼性羥基和強酸性羥基相對高度分別為96%和52%,且強鹼性羥基和強酸性羥基的峰高比為1:0.68。
對比例6
將氫氧化鋯粉(比表面積為260m2/g)、田菁粉按照100g:2.5g比例混合均勻後,加入70mL的稀硝酸(濃度為2重量%),並再次攪拌均勻擠條成型。經過在100℃下乾燥5h和在500℃下焙燒3h步驟,製備得到粒徑為1.6mm的氧化鋯載體D6。載體D6的比表面積為108m2/g。
實施例5
將氫氧化鋯粉(同對比例6)、田菁粉按照100g:2.5g比例混合均勻後,首先加入含4.88g的SiO2的矽溶膠攪拌均勻,而後加入70mL的稀硝酸(濃度為2重量%),並再次攪拌均勻並擠條成型。載體經過在100℃下乾燥5h和在550℃下焙燒3h後得到氧化鋯載體,焙燒後的載體以孔飽和浸漬法引入4.81g的硝酸鎂,然後經過120℃乾燥3h和500℃焙燒4h,得到Sup5載體。載體Sup5的比表面積為102m2/g。
利用紅外光譜法考察上述對比例6和實施例5獲得的載體的羥基振動特徵發現,Sup5中載體的強鹼性羥基和強酸性羥基的相對高度分別為80%和75%,且強鹼性羥基和強酸性羥基的峰高比為1:1.01;而載體D6中,強鹼性羥基和強酸性羥基的峰高比為1:1.35。
實施例1-1
以Sup1為載體製備含Ni和W的催化劑Cat-Sup1,其中表面W原子濃度為3.5個原子/nm2,Ni/(Ni+W)原子比為0.3。催化劑的活性評價分別在固定床加氫脫硫評價裝置上進行。原料油性質如下:密度為0.8457g/cm3,硫含量為1.1wt%,氮含量為223ppm。反應條件為:溫度為325℃,壓力為3.2MPa,重時空速為2h-1,氫油比為300v/v。反應在120h和240h時硫的轉化率以及催化劑的脫硫轉化率損失如表1所示,其中脫硫轉化率損失為兩個時段催化劑脫硫率差值。
對比例1-1
使用與實施例1-1相同重量的Ni、W金屬活性組分並採用與實施例1-1相同的方法製備含Ni和W的催化劑Cat-D1,不同的是,載體Sup1由對比例1製得的純氧化鋁載體D1代替。然後採用與實施例1-1相同的方法進行加氫脫硫活性評價,結果如表1所示。
實施例2-1
以鹼式碳酸鈷、氧化鉬、磷酸和乙二醇為原料,以Sup2為載體,通過飽和浸漬法製備得到Cat-Sup2催化劑。載體表面Mo原子濃度為3.2原子/nm2,Co/(Co+Mo)原子比為0.32。然後採用與實施例1-1相同的方法進行加氫脫硫活性評價,結果如表1所示。
對比例2-1
使用與實施例2-1相同重量的Co、Mo金屬活性組分並採用與實施例2-1相同的方法製備含Co和Mo的催化劑Cat-D2,不同的是,載體Sup2由對比例2製得的純氧化鋁載體D2代替。然後採用與實施例1-1相同的方法進 行加氫脫硫活性評價,結果如表1所示。
實施例3-1
以鹼式碳酸鎳、氧化鉬、偏鎢酸銨、磷酸和檸檬酸為原料,以Sup3為載體,通過飽和浸漬法製備得到Cat-Sup3催化劑。載體表面W原子濃度為2.8個原子/nm2,Mo原子濃度為0.4個原子/nm2,Ni/(Ni+Mo+W)原子比為0.29。然後採用與實施例1-1相同的方法進行加氫脫硫活性評價,結果如表1所示。
對比例3-1
使用與實施例3-1相同重量的Ni、W和Mo金屬活性組分並採用與實施例3-1相同的方法製備含Ni和W和Mo的催化劑Cat-D3,不同的是,載體Sup3由對比例3製得的純氧化鋁載體D3代替。然後採用與實施例1-1相同的方法進行加氫脫硫活性評價,結果如表1所示。
實施例4-1
使用與實施例3-1相同重量的Ni、W和Mo金屬活性組分並採用與實施例3-1相同的方法製備含Ni和W和Mo的催化劑Cat-Sup4,不同的是,載體Sup3由實施例4製得的載體代替Sup4。然後採用與實施例1-1相同的方法進行加氫脫硫活性評價,結果如表1所示。
對比例4-1
使用與實施例3-1相同重量的Ni、W和Mo金屬活性組分並採用與實施例3-1相同的方法製備含Ni和W和Mo的催化劑Cat-D4,不同的是,載體Sup3由對比例4製得的載體D4替代。然後採用與實施例1-1相同的方法進 行加氫脫硫活性評價,結果如表1所示。
對比例5-1
使用與實施例1-1相同重量的Ni、W金屬活性組分並採用與實施例1-1相同的方法製備含Ni和W的催化劑Cat-D5,不同的是,載體Sup1由對比例5製得的Cat-D5載體代替。然後採用與實施例1-1相同的方法進行加氫脫硫活性評價,結果如表1所示。
對比例6-1
使用與實施例1-1相同重量的Ni、W金屬活性組分並採用與實施例1-1相同的方法製備含Ni和W的催化劑Cat-D6,不同的是,載體Sup3由對比例6製得的載體D6代替。然後採用與實施例1-1相同的方法進行加氫脫硫活性評價,結果如表1所示。
實施例5-1
使用與實施例1-1相同重量的Ni、W金屬活性組分並採用與實施例1-1相同的方法製備含Ni和W的催化劑Cat-Sup5,不同的是,載體Sup3由實施例5製得的載體Sup5代替。然後採用與實施例1-1相同的方法進行加氫脫硫活性評價,結果如表1所示。
實施例6-1
採用與實施例1-1相同的方法製備含Ni和W的催化劑Cat-Sup6,不同的是,調整原料的用量,使得載體表面W原子濃度為3個原子/nm2,Ni/(Ni+W)原子比為0.1。然後採用與實施例1-1相同的方法進行加氫脫硫活性評價,結果如表1所示。
表1
從表1的結果可以看出,採用本發明提供的載體並配以特定量的金屬活性成分製得的催化劑的脫硫活性得到了提高,失活速度得到了抑制。由此可以看出,載體的羥基經過處理後,催化劑的脫硫活性得到了提高,失活速率得到了降低。
以上詳細描述了本發明的優選實施方式,但是,本發明並不限於上述實施方式中的具體細節,在本發明的技術構思範圍內,可以對本發明的技術方案進行多種簡單變型,這些簡單變型均屬於本發明的保護範圍。
另外需要說明的是,在上述具體實施方式中所描述的各個具體技術特徵,在不矛盾的情況下,可以通過任何合適的方式進行組合。為了避免不必要的重複,本發明對各種可能的組合方式不再另行說明。
此外,本發明的各種不同的實施方式之間也可以進行任意組合,只要其不違背本發明的思想,其同樣應當視為本發明所公開的內容。