一種左旋艾普拉唑鈉化合物及其藥物組合物和製備方法與流程
2023-04-26 19:02:21 3
本發明屬於治療胃病的藥物領域,特別涉及一種左旋艾普拉唑鈉化合物及其藥物組合物和製備方法。
背景技術:
艾普拉唑(ilaprazole),化學名:5-(1-氫-吡咯-1-基)-2[[(4-甲氧基-3-甲基)-2-吡啶基]-甲基-亞磺醯基-1-氫-苯並咪唑。其是一種新型質子泵抑制劑,由韓國一洋藥品株式會社開發,主要用於十二指腸潰瘍、胃潰瘍、反流性或腐蝕性食管炎等的治療。
與其他質子泵抑制劑例如奧美拉唑等相比,具有起效快、抑酸效果好、作用時間長等優勢,當時該化合物及其鈉鹽穩定性差,為了提高該化合物的穩定性,現有技術研究出了多種該化合物及其鈉鹽的晶型,例如cn101687878a公開了艾普拉唑的a、b、e、f、i晶型及其製備方法,cn103172618a公開了艾普拉唑的m晶型,又如cn102746275a公開了一種結晶形式的艾普拉唑鈉,又如cn105461692a公開了一種艾普拉唑鈉的c晶型,等。雖然現有技術公開了多種艾普拉唑及其鈉鹽的晶型,但是以上公開的晶型穩定性仍較差。
此外,由於艾普拉唑鈉對酸不穩定,所以常將其製成凍乾粉或腸溶片,例如cn105769777a公開的一種艾普拉唑鈉的凍乾粉針劑,及cn103705476a公開的一種艾普拉唑凍乾粉針劑,cn102552190公開的一種腸溶片,以上公開的製劑解決了艾普拉唑鈉對酸不穩定的技術問題,當上述限定的凍乾粉的穩定性較低,並且凍幹保護後化合物的有關物質低。
技術實現要素:
為了解決上述技術問題,本發明提供了一種左旋艾普拉唑鈉化合物及其藥物組合物和製備方法,該左旋艾普拉唑鈉化合物的穩定性顯著提高。
本發明具體方案如下:
本發明提供一種左旋艾普拉唑鈉化合物,該化合物以2θ角度表示的x-射線衍射在7.6°、14.3°、19.8°、24.1°處有特徵峰。
進一步的改進,該化合物以2θ角度表示的x-射線衍射還在9.6°、11.5°、14.1°、18.0°、25.3°、31.9°、35.1°處有特徵峰。
本發明另一方面提供一種製劑,該製劑包括左旋艾普拉唑鈉化合物和輔料,該製劑為凍乾粉或腸溶片,該左旋艾普拉唑鈉化合物和輔料的重量份數比為1:1.5-3。
進一步的改進,該製劑為凍乾粉,其中,輔料包括如下重量份的各成分:
卡波姆1-1.8
穀氨酸鈉2.5-6。
本發明在凍乾粉中加入卡波姆和穀氨酸鈉的混合物,兩者在水溶液中能夠形成一層凝膠起到保護化合物的作用,進而提高了凍乾粉中左旋艾普拉唑鈉化合物的穩定性,提高了凍乾粉的穩定性。
進一步的改進,輔料還包括重量份為1.2-3.5份的凍幹保護劑。進一步的改進,所該凍幹保護劑各成分及重量份如下:
海藻糖1-3
維生素c0.2-0.5。
本發明進一步選擇海藻糖和維生素c的混合物作為凍幹保護劑,提高了凍乾粉的有關物質,進而提高了凍乾粉的整體療效。
進一步的改進,輔料還包括如下重量份的各成分:
聚丙烯酸鈉1-3
十二烷基磺酸鈉0.5-1。
本發明在凍乾粉中加入聚丙烯酸鈉和十二烷基磺酸鈉的混合物可以與化合物形成微球,不但提高了化合物的溶解性及穩定性,使得化合物具有緩釋特性,延長凍乾粉的作用時間。
進一步的改進,該凍乾粉各成分的重量份數如下:
本發明另一方面提供了左旋艾普拉唑鈉化合物的製備方法,該方法包括如下步驟:
1)將1g左旋艾普拉唑鈉溶於50ml溶液a中,製得混合液;其中溶液a為體積比為11.5:1-1.5的四氫呋喃和乙醇的混合物,攪拌,轉速為100r/min;
2)將混合液濃縮至原體積的2/3後,向濃縮後的混合液中加入5-10ml乙醚,然後再緩慢滴加2-3ml濃度為0.02mol/l的碳酸氫鈉水溶液,靜置,析晶。
本發明另一方面還提供了凍乾粉的製備方法,該方法包括如下步驟:
s1:將卡波姆加入水中,於研缽中研勻後,將穀氨酸鈉加入研缽中攪拌均勻,製得凝膠液;
s2:將左旋艾普拉唑鈉化合物、凝膠液、聚丙烯酸鈉溶於水中,製得水溶液;
s3:將十二烷基磺酸鈉溶於乙醇中,製得乙醇溶液;
s4:將步驟s3製得的乙醇溶液加入到步驟s2製得的水溶液中,1600r·min-1攪拌,形成乳液;
s5:控制乳液溫度低於20℃下,繼續攪拌,使乙醇揮發至微球固化,得微球沉澱物,離心後加入凍幹保護劑,攪拌30min,冷凍乾燥,即得。
本發明的有益效果在於,本發明提供一種左旋艾普拉唑鈉化合物,該化合物具有新的結晶形式,其穩定性較現有艾普拉唑鈉的晶型形式顯著提高;此外,本發明還提供了包括左旋艾普拉唑鈉化合物的凍乾粉和腸溶片,該凍乾粉具有穩定性高、緩釋等效果,療效優於現有的凍乾粉或腸溶片。
附圖說明
圖1為左旋艾普拉唑鈉化合物的x-射線衍射圖;
圖2凍乾粉的體外釋放度圖。
具體實施例方式
實施例1一種左旋艾普拉唑鈉化合物
該化合物以2θ表示的x-射線衍射在7.6°、14.3°、19.8°、24.1°處有特徵峰,如圖1所示,以上各特徵峰的相對強度如下所示:
實施例2一種左旋艾普拉唑鈉化合物
該化合物以2θ表示的x-射線衍射在7.6°、9.6°、11.5°、14.1°、14.3°、18.0°、19.8°、24.1°、25.3°、31.9°、35.1°處有特徵峰,如圖1所示,以上各特徵峰的相對強度如下所示:
實施例3凍乾粉
凍乾粉各成分的用量為:
實施例1的化合物1.75g
卡波姆1g
穀氨酸鈉2.5g。
實施例4凍乾粉
凍乾粉各成分的用量為:
實施例2的化合物2.4g
卡波姆1.8g
穀氨酸鈉5.4g
製備方法:
s1:將1.8g卡波姆加入30ml水中,於研缽中研勻後,將5.4g穀氨酸鈉加入研缽中攪拌均勻,製得凝膠液;
s2:將2.4g左旋艾普拉唑鈉化合物溶解於步驟s1獲得的凝膠液中,攪拌至完全溶解,再繼續攪拌30min,冷凍乾燥,即得。
實施例5凍乾粉
凍乾粉各成分的用量為:
實施例6凍乾粉
凍乾粉各成分的用量為:
製備方法:
s1:將1.2g卡波姆加入50ml水中,於研缽中研勻後,將3.6g穀氨酸鈉加入研缽中攪拌均勻,製得凝膠液;
s2:將2.84g左旋艾普拉唑鈉化合物溶解於步驟s1獲得的凝膠液中,攪拌至完全溶解,加入2g海藻糖和0.3g維生素c,再繼續攪拌30min,冷凍乾燥,即得。
實施例7凍乾粉
凍乾粉各成分的用量為:
實施例8凍乾粉
凍乾粉各成分的用量為:
實施例9
凍乾粉各成分的用量為:
製備方法:
s1:將1.5g卡波姆加入50ml水中,於研缽中研勻後,將4.5g穀氨酸鈉加入研缽中攪拌均勻,製得凝膠液;
s2:將5g左旋艾普拉唑鈉化合物、凝膠液、2g聚丙烯酸鈉溶於100ml水中,製得水溶液;
s3:將十0.75二烷基磺酸鈉溶於10ml乙醇中,製得乙醇溶液;
s4:將步驟s3製得的乙醇溶液加入到步驟s2製得的水溶液中,1600r·min-1攪拌,形成乳液;
s5:控制乳液溫度低於20℃下,繼續攪拌,使乙醇揮發至微球固化,得微球沉澱物,離心後加入2.5g海藻糖和0.25g維生素c,攪拌30min,冷凍乾燥,即得。
對照例1凍乾粉
凍乾粉各成分的用量為:
實施例1的化合物1.75g
卡波姆1g
依地酸二鈉2.5g
按照實施例4的方法製備。
對照例2凍乾粉
凍乾粉各成分的用量為:
實施例1的化合物1.75g
羧甲基纖維素鈉1g
穀氨酸鈉2.5g
按照實施例4的方法製備。
對照例3凍乾粉
凍乾粉各成分的用量為:
按照實施例9的方法製備。
對照例4凍乾粉
凍乾粉各成分的用量為:
按照實施例9的方法製備。
實驗例1化合物穩定性實驗
以下通過對比實驗來說明本發明與現有技術中的艾普拉唑鈉晶型的穩定性差異。
1.高溫影響因素實驗
分別取實施例2、對照例1(cn10546169a2公開的c晶型)、對照例2(cn102746277a公開的結晶形式的艾普拉唑鈉)、對照例3(cn104922080a公開的艾普拉唑鈉晶體)、對照例4(cn106749191公開的艾普拉唑的ⅱ型結晶)、對照例5(cn105147680a公開的一種抗胃酸分泌的藥物艾普拉唑鈉組合物中艾普拉唑鈉晶體)的艾普拉唑鈉或艾普拉唑晶體,在溫度60℃,相對溼度75%的條件下放置10天,分別在第0、5、10天取樣觀察其外觀、色澤並測定雜質含量,結果見表1。
表1高溫影響因素實驗結果
結果表明,本發明提供的左旋艾普拉唑鈉的晶型在高溫條件下放置10天,從性狀、外觀上看沒有變化,有關物質和含量的變化較對照例4和對照例5小,且明顯好於對照例1-3。
2.高溼影響因素實驗
分別取實施例2、對照例1(cn10546169a2公開的c晶型)、對照例2(cn102746277a公開的結晶形式的艾普拉唑鈉)、對照例3(cn104922080a公開的艾普拉唑鈉晶體)、對照例4(cn106749191公開的艾普拉唑的ⅱ型結晶)、對照例5(cn105147680a公開的一種抗胃酸分泌的藥物艾普拉唑鈉組合物中艾普拉唑鈉晶體)的艾普拉唑鈉或艾普拉唑晶體,在溫度25℃,相對溼度92.5%的條件下放置10天,分別在第0、5、10天取樣觀察其外觀、色澤並測定雜質含量,結果見表2。
表2高溼影響因素實驗結果
結果表明,本發明提供的左旋艾普拉唑鈉的晶型在高溼條件下放置10天,從性狀、外觀上看沒有變化,有關物質和含量的變化較對照例4和對照例5小,且明顯好於對照例1-3。
3.強光影響因素實驗
分別取實施例2、對照例1(cn10546169a2公開的c晶型)、對照例2(cn102746277a公開的結晶形式的艾普拉唑鈉)、對照例3(cn104922080a公開的艾普拉唑鈉晶體)、對照例4(cn106749191公開的艾普拉唑的ⅱ型結晶)、對照例5(cn105147680a公開的一種抗胃酸分泌的藥物艾普拉唑鈉組合物中艾普拉唑鈉晶體)的艾普拉唑鈉或艾普拉唑晶體,在照度為4000-5000lx的日光燈的光照箱內放置10天,分別在第0、5、10天取樣觀察其外觀、色澤並測定雜質含量,結果見表3。
表3強光影響因素實驗結果
結果表明,本發明提供的左旋艾普拉唑鈉的晶型在強光條件下放置10天,從性狀、外觀上看沒有變化,有關物質和含量的變化較對照例4和對照例5小,且明顯好於對照例1-3。
實驗例2凍乾粉穩定性實驗
1加速試驗
取本發明實施例3、及對照例1和2的凍乾粉,均在40℃±2℃下,相對溼度為75%±5%的條件下放置6個月,在試驗期間1個月、2個月、3個月、6個月末分別取樣一次,按中國藥典中的規定,檢測凍乾粉的性狀、左旋艾普拉唑鈉的含量(標示量%)、有關物質(%,總雜),結果見表4。
表4凍乾粉的加速試驗結果
從表中可看出本發明實施例3所提供的凍乾粉,經加速試驗結果可知,放置6個月後,所述凍乾粉的性狀、含左旋艾普拉唑鈉的量、有關物質均未發生明顯的變化;而對照例1-3的凍乾粉放置6個月後,凍乾粉發黃,且左旋艾普拉唑鈉的含量顯著下降,有關物質顯著升高;表明本發明的凍乾粉與對照例1-2相比,穩定性顯著提高。
1.2長期試驗
取本發明實施例3及對照例1-2的凍乾粉,均於溫度25℃±2℃下,相對溼度為60%±10%的條件下放置12個月,每3個月取樣一次,分別於0個月、3個月、6個月、9個月、12個月、24個月取樣,檢測凍乾粉的性狀、左旋艾普拉唑鈉的含量(標示量%)及有關物質(%,總雜),結果見表5。
表5凍乾粉的長期試驗結果
從表中可看出本發明實施例3所提供的凍乾粉,經長期試驗結果可知,放置24個月後,所述凍乾粉的性狀、含左旋艾普拉唑鈉的量、有關物質均未發生明顯的變化;而對照例1-2的凍乾粉放置24個月後,凍乾粉發黃,並且左旋艾普拉唑鈉的含量顯著下降,有關物質升高;表明本發明的凍乾粉與對照例1-2的凍乾粉相比,穩定性顯著提高。
實驗例3不同凍幹保護劑對凍乾粉的影響
本試驗選擇不同的凍幹保護劑,其餘的成分與實施例5的相同,製成不同的凍乾粉,對凍乾粉的包封率、外觀、再分散時間等因素進行考察,考察結果見表6。
表6不同凍幹保護劑對凍乾粉的影響結果
從表中可以看出,當選擇海藻糖和維生素c的混合物作為凍幹保護劑時,製得的凍乾粉的外觀為白色、飽滿,包封率和再分散時間好,其餘的凍幹保護劑外觀不好,且有萎縮現象,分散時間長,包封率低。
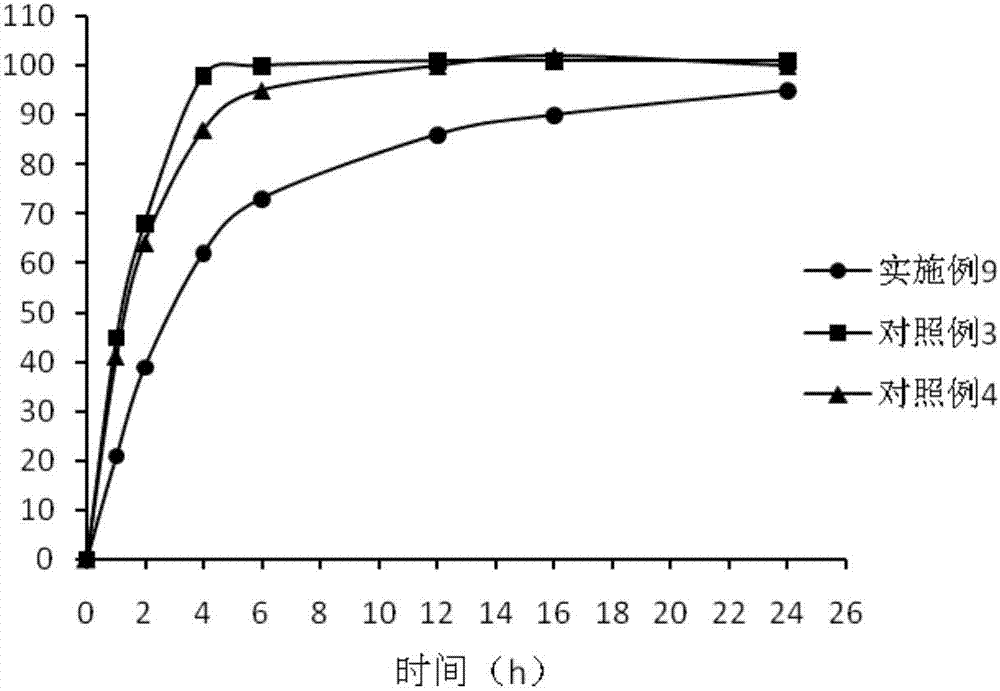
實驗例4體外釋放度測定試驗
藥物釋放度檢測:參照2015版《中國藥典》附錄xixd體外藥物釋放度檢查。
分別取以上實施例9和對照例3-4的凍乾粉,置藥物溶出儀中,於1h、2h、4h、6h、12h、16h、24h分別取樣,用高效液相色譜法檢測溶出百分率,並計算藥物的累積釋放百分率,結果見圖2。
從圖中可以看出實施例9的凍乾粉,在24h內緩慢釋放。