一種用於抗腫瘤藥物奈拉濱粉針劑組合物的奈拉濱化合物的製作方法
2023-12-10 06:12:42 1
本發明屬於醫藥技術領域,涉及一種抗腫瘤藥物奈拉濱化合物。
背景技術:
奈拉濱化學名為2-氨基-9-β-D-阿拉伯呋喃糖基-6-甲氧基-9H-嘌呤,是脫氧鳥苷類似物9-β-D-阿糖呋喃糖鳥嘌呤(ara-G)的前體藥,2005年已被批准上市,可用於治療T細胞急性淋巴細胞性白血病(T-ALL)和T細胞淋巴母細胞性淋巴瘤(T-LBL)。結構式如下:
奈拉濱為白色或類白色結晶性粉末,微溶於水,在水中溶解度約為8-9mg/ml(25℃,pH=4-10)。由於奈拉濱在水中的溶解性較差,這在一定程度上限制了其在醫藥上的應用。
「奈拉濱的合成研究進展」[梁平,尹先清,李衛佳.精細化工中間體,2008,38(5):8-10]、CN101348511A、CN101092441A均公開了一種將奈拉濱加入甲醇混合加熱至回流,攪拌使物料溶解,趁熱過濾,濾液濃縮,攪拌下加入無水乙醇、乙醚,冷卻放置析晶的方法。
專利 CN101348511A公開了奈拉濱的合成及精製,其中奈拉濱結晶的製備方法包括:奈拉濱加入甲醇混合加熱至回流,溶解後趁熱過濾,濾液濃縮,攪拌下加入乙醇或乙醚,放置冷卻析晶,過濾,用無水乙醇洗滌,乾燥,得到奈拉濱結晶。
專利CN103172687A公開了一種奈拉濱結晶化合物及其製備方法,其製備方法為:取奈拉濱粗品,投入無水甲醇中,加熱至回流;奈拉濱粗品溶解後加入活性炭回流,趁熱過濾,降至室溫,冰鹽浴降溫,析晶,抽濾,冰甲醇洗滌濾餅得到奈拉濱結晶化合物。
專利CN103191051A公開了一種奈拉濱注射液組合物及其製備方法,其藥物活性成分為奈拉濱和氯化鈉,其中,所述奈拉濱為一種奈拉濱結晶化合物,該結晶化合物採用粉末X-射線衍射測定法測定,得到的X射線粉末衍射圖譜如圖5所示。
本領域技術人員都知道,藥物的多晶形已經成為藥物研究過程和藥品生產質量控制及檢測過程中必不可少的重要組成部分。對藥物多晶形的研究有助於新藥化合物生物活性的選擇,有助於提高生物利用度,增進臨床療效,有助於藥物給藥途徑的選擇與設計,以及藥物製劑工藝參數的確定,從而提高藥品生產質量。同一藥物晶形不同,其生物利用度可能差異顯著。同一種藥物,某些晶形可能比其他晶形具備更高的生物活性。
本發明經過大量的研究,得到了一種不同於現有技術的奈拉濱新晶體化合物,通過試驗驗證,發現該新晶體化合物純度高,流動性及水溶性好,穩定性高,不易吸溼,臨床應用安全可靠,利用該新晶體化合物製得的粉針劑組合物不僅純度高、雜質含量低、澄明度好,而且能保證生產中的分裝效率、裝量差異小,不良反應發生率大大降低,穩定性更好。
技術實現要素:
本發明的發明目的在於提供一種抗腫瘤藥物奈拉濱化合物。
為了完成本發明的目的,採用的技術方案為:
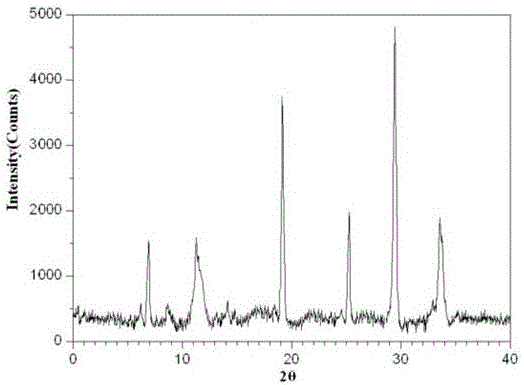
本發明公開一種抗腫瘤藥物奈拉濱化合物,所述的奈拉濱為晶體,使用Cu-Kα射線測量得到的X-射線粉末衍射圖如圖1所示。
本發明公開上述抗腫瘤藥物奈拉濱化合物的製備方法,包括如下步驟:
(1)將奈拉濱粗品加入有機溶劑1中,加熱至70-80℃,攪拌0.5-1.5小時;
(2)待奈拉濱粗品完全溶解,加入活性炭,繼續升溫至回流,保溫10-30分鐘,趁熱過濾,濾液濃縮;
(3)攪拌下加入有機溶劑2,放置冷卻至室溫;
(4)繼續放置1-3小時,冰浴降溫至-15℃--5℃析晶,養晶6-10小時,過濾,用少量冷卻到0℃的無水乙醇洗滌晶體至少1次,乾燥,得奈拉濱晶體。
進一步的,步驟(1)中所述奈拉濱粗品與有機溶劑1的質量體積比為1g:10-12ml。
進一步的,步驟(2)中所述活性炭的質量為奈拉濱粗品質量的0.1-0.3%。
進一步的,步驟(3)中所述有機溶劑2與有機溶劑1的體積比為1:0.25-0.50。
進一步的,步驟(4)中所述乾燥為減壓乾燥,乾燥溫度控制在50℃以下,壓力控制在5-15mmHg,乾燥時間為10-15小時。
進一步的,步驟(1)中所述有機溶劑1為乙醇或乙腈或二者1:1的混合物,步驟(3)中所述有機溶劑2為異丙醇。
本發明中,所述的奈拉濱粗品可以是採用現有技術的方法所公開的奈拉濱的合成方法製備得到的奈拉濱,也可以是市售的奈拉濱原料藥。
本發明還公開一種抗腫瘤藥物奈拉濱粉針劑組合物,所述的粉針劑組合物的組成為:奈拉濱1重量份,精氨酸0.1-0.3重量份;所述的奈拉濱為晶體,使用Cu-Kα射線測量得到的X-射線粉末衍射圖如圖1所示。
進一步的,所述的粉針劑的組成為:奈拉濱1重量份,精氨酸0.2重量份。
進一步的,所述的粉針劑組合物的製備方法包括以下步驟:
(1)按比例稱取奈拉濱晶體和精氨酸,充分混合;
(2)分裝至滅菌後的西林瓶中並加塞。
固體化學藥物的多晶型現象是一種普遍物質存在的自然現象,這種現象是指一種固體化學藥物可以存在2種或2種以上晶型狀態,又稱為物質的多晶型狀態,物質的多晶型狀態也稱為「同質異晶」現象。同質異晶的固體物質雖然其化學本質是相同的,但其理化性質可能是不同的。對於理化性質不同的「同質異晶藥物」,在臨床上也可以表現出不同防治疾病的療效,直接影響藥物的應用和臨床效果。
本發明進一步通過實施例和實驗例,發現本發明的奈拉濱晶體化合物較現有技術相比提高了水溶性,且生物利用度高、穩定性好、收率高、純度高,在一定程度上拓寬了其在醫藥上的應用。
與現有技術相比,本發明具有如下優點:
(1)本發明所提供的奈拉濱晶體是一種不同於現有技術的奈拉濱晶體,該晶體能顯著提高奈拉濱在水中的溶解度,且流動性好,不易吸溼。
(2)本發明所提供的奈拉濱晶體具有生物利用度高、藥效顯著、穩定性高、收率高,純度高等優點,有助於藥物給藥途徑的選擇設計以及藥物製劑工藝參數的確定,從而提高藥品生產質量。
(3)本發明所提供的奈拉濱晶體化合物的製備方法操作簡便,是一種經濟可行、適合工業化大生產的方法。
(4)利用奈拉濱新晶體化合物製得的粉針劑組合物不僅純度高、雜質含量低、澄明度好,而且能保證生產中的分裝效率、裝量差異小,不良反應發生率大大降低,穩定性更好。
附圖說明
圖1為本發明實施例1製備的奈拉濱晶體的X-射線粉末衍射圖譜。
圖2為本發明實施例1製備的奈拉濱晶體化合物的熱分析圖譜。
具體實施方式
下面通過具體實施例對本發明的發明內容作進一步詳細的說明,但並不因此而限定本發明的內容。
實施例1:奈拉濱晶體的製備
(1)將10g奈拉濱粗品加入110ml乙醇和乙腈(1:1)的混合物中,加熱至75℃,攪拌1小時;
(2)待奈拉濱粗品完全溶解,加入活性炭0.02g,繼續升溫至回流,保溫20分鐘,趁熱過濾,濾液濃縮;
(3)攪拌下加入330ml異丙醇,放置冷卻至室溫;
(4)繼續放置2小時,冰浴降溫至-10℃析晶,養晶8小時,過濾,用少量冷卻到0℃的無水乙醇洗滌晶體至少1次,減壓乾燥(乾燥溫度控制在50℃以下,壓力控制在10mmHg,乾燥時間為12小時),得奈拉濱晶體化合物9.875g,收率98.75%,純度99.99%。
所製得的奈拉濱晶體化合物以2θ±0.2°衍射角表示的X-射線粉末衍射圖譜在6.9°、8.9°、11.5°、19.1°、25.6°、29.7°、33.9°處顯示有特徵衍射峰,使用Cu-Kα射線測量得到如圖1所示的X射線粉末衍射圖譜,高效液相色譜測定其純度為99.9%。與現有技術各晶型的X-射線粉末衍射圖譜進行對比,明顯發現本發明奈拉濱晶型化合物不同於現有技術。
採用卡爾·費休水分測定法測定KF為0.39%,所以本發明奈拉濱晶型化合物不含結晶水。
將所製得的奈拉濱晶體化合物採用美國Perkin-Elmer 公司PE Pyris Diamond TG 熱分析儀得到的熱重分析圖如圖2所示。
實施例2:奈拉濱晶體的製備
(1)將10g奈拉濱粗品加入105ml乙醇和乙腈(1:1)的混合物中,加熱至78℃,攪拌0.8小時;
(2)待奈拉濱粗品完全溶解,加入活性炭0.025g,繼續升溫至回流,保溫15分鐘,趁熱過濾,濾液濃縮;
(3)攪拌下加入420ml異丙醇,放置冷卻至室溫;
(4)繼續放置2.5小時,冰浴降溫至-15℃析晶,養晶7小時,過濾,用少量冷卻到0℃的無水乙醇洗滌晶體至少1次,減壓乾燥(乾燥溫度控制在50℃以下,壓力控制在12mmHg,乾燥時間為13小時),得奈拉濱晶體化合物9.856g,收率98.56%,純度99.99%。
所製得的奈拉濱晶體化合物使用Cu-Kα射線測量得到的X-射線粉末衍射譜圖與實施例1相似;採用美國Perkin-Elmer 公司PE Pyris Diamond TG 熱分析儀得到的熱重分析圖與實施例1相似。
實施例3:奈拉濱晶體的製備
(1)將10g奈拉濱粗品加入115ml乙醇和乙腈(1:1)的混合物中,加熱至72℃,攪拌1.2小時;
(2)待奈拉濱粗品完全溶解,加入活性炭0.015g,繼續升溫至回流,保溫25分鐘,趁熱過濾,濾液濃縮;
(3)攪拌下加入230ml異丙醇,放置冷卻至室溫;
(4)繼續放置1.5小時,冰浴降溫至-5℃析晶,養晶9小時,過濾,用少量冷卻到0℃的無水乙醇洗滌晶體至少1次,減壓乾燥(乾燥溫度控制在50℃以下,壓力控制在8mmHg,乾燥時間為11小時),得奈拉濱晶體化合物9.849g,收率98.49%,純度99.99%。
所製得的奈拉濱晶體化合物使用Cu-Kα射線測量得到的X-射線粉末衍射譜圖與實施例1相似;採用美國Perkin-Elmer 公司PE Pyris Diamond TG 熱分析儀得到的熱重分析圖與實施例1相似。
實施例4:奈拉濱晶體的製備
(1)將10g奈拉濱粗品加入100ml乙醇中,加熱至70℃,攪拌0.5小時;
(2)待奈拉濱粗品完全溶解,加入活性炭0.01g,繼續升溫至回流,保溫30分鐘,趁熱過濾,濾液濃縮;
(3)攪拌下加入350ml異丙醇,放置冷卻至室溫;
(4)繼續放置1小時,冰浴降溫至-12℃析晶,養晶10小時,過濾,用少量冷卻到0℃的無水乙醇洗滌晶體至少1次,減壓乾燥(乾燥溫度控制在50℃以下,壓力控制在5mmHg,乾燥時間為15小時),得奈拉濱晶體化合物9.827g,收率98.27%,純度99.85%。
所製得的奈拉濱晶體化合物使用Cu-Kα射線測量得到的X-射線粉末衍射譜圖與實施例1相似;採用美國Perkin-Elmer 公司PE Pyris Diamond TG 熱分析儀得到的熱重分析圖與實施例1相似。
實施例5:奈拉濱晶體的製備
(1)將10g奈拉濱粗品加入120ml乙腈中,加熱至80℃,攪拌1.5小時;
(2)待奈拉濱粗品完全溶解,加入活性炭0.03g,繼續升溫至回流,保溫10分鐘,趁熱過濾,濾液濃縮;
(3)攪拌下加入300ml異丙醇,放置冷卻至室溫;
(4)繼續放置3小時,冰浴降溫至-8℃析晶,養晶6小時,過濾,用少量冷卻到0℃的無水乙醇洗滌晶體至少1次,減壓乾燥(乾燥溫度控制在50℃以下,壓力控制在15mmHg,乾燥時間為10小時),得奈拉濱晶體化合物9.832g,收率98.32%,純度99.99%。
所製得的奈拉濱晶體化合物使用Cu-Kα射線測量得到的X-射線粉末衍射譜圖與實施例1相似;採用美國Perkin-Elmer 公司PE Pyris Diamond TG 熱分析儀得到的熱重分析圖與實施例1相似。
製劑實施例1:奈拉濱粉針劑的製備:
組成為:本發明製備的奈拉濱晶體1重量份,精氨酸0. 1重量份。
製備方法為:
(1)按比例稱取奈拉濱晶體和精氨酸,充分混合;
(2)分裝至滅菌後的西林瓶中並加塞。
製劑實施例2:奈拉濱粉針劑的製備:
組成為:本發明製備的奈拉濱晶體1重量份,精氨酸0.2重量份。
製備方法為:
(1)按比例稱取奈拉濱晶體和精氨酸,充分混合;
(2)分裝至滅菌後的西林瓶中並加塞。
製劑實施例3:奈拉濱粉針劑的製備:
組成為:本發明製備的奈拉濱晶體1重量份,精氨酸0. 3重量份。
製備方法為:
(1)按比例稱取奈拉濱晶體和精氨酸,充分混合;
(2)分裝至滅菌後的西林瓶中並加塞。
試驗品1:本發明實施例1所製備的奈拉濱晶體化合物。
試驗品2:本發明實施例4所製備的奈拉濱晶體化合物。
對照品1:按照CN101348511A實施例19的方法製得奈拉濱晶體化合物。
對照品2:按照CN103172687A實施例1的方法製得奈拉濱晶體化合物。
對照品3:市售奈拉濱
實驗例1:流動性試驗
本實驗例通過測定樣品的休止角來評價樣品的流動性,具體方法如下:取樣品顆粒,從固定的小漏鬥中流入圓形的表面皿中,直到得到最高的圓椎體,量取圓錐體高度H和半徑R,按tanα=H/R計算休止角α,結果見表1,休止角越大,流動性越差。
表1 流動性試驗結果
從表1可知,與現有技術中的奈拉濱相比,本發明所製備的奈拉濱晶體化合物流動性顯著提高,有利於製劑的製備,溶出度、生物利用度的提高。
實驗例2:溶解性試驗
在帶有恆溫夾套的小容量瓶中加入適量的蒸餾水,在25℃下加入奈拉濱至不再溶解為止,啟動電磁攪拌器,恆溫下持續攪拌,在實驗過程中體系始終處於兩相共存的狀態,70分鐘後體系的液相中奈拉濱的濃度即為該溫度下的溶解度。在2小時後進行取樣分析,取相鄰兩次結果相近的平均值作為實驗測定值,取樣前,為了使固液充分分離,停止攪拌後,未溶的奈拉濱沉澱到小容量瓶的底部,用注射器抽取少量上部清液,用0.45微米的濾頭過濾,從濾液中取樣品,通過HPLC測出奈拉濱的含量(濃度(mg/ml))。結果見表2。
表2 室溫下本發明奈拉濱新晶體化合物與現有技術晶型的水溶性對比
從表2可以看出,本發明奈拉濱新晶體化合物的水溶性與現有技術相比,有顯著提高。
實驗例3:吸溼性試驗
將各樣品開口置潔淨培養皿中,攤成≤5mm厚的薄層,各兩份,分別放入恆溼密閉容器中,在25℃分別於相對溼度75%與92.5%的條件下放置10天,於第5天和第10天取樣,以費歇爾法測定各樣品的水分含量,試驗結果與0天比較,實驗結果見表3。
表3 吸溼性試驗結果
從上表可以看出,本發明製備的奈拉濱晶體化合物在高溼度條件下基本不吸溼,其在高溼環境下的穩定性明顯優於採用現有技術的奈拉濱。
實驗例4:影響因素試驗
將試驗品和對照品模擬上市包裝,在高溫60℃、光照4500Lx、高溼(RH90%±5%)條件下放置10天,按穩定性重點考察項目進行檢測,與0天樣品比較,結果見表4。
有關物質測定方法:
照高效液相色譜法(中國藥典2005年版二部附錄VD)測定。
色譜條件與系統適用性實驗:以十八烷基矽烷鍵合矽膠為填充劑;以0.02mol/L磷酸二氫鈉的水溶液:甲醇(83:17)為流動相;檢測波長:210nm。奈拉濱峰理論塔板數應不低於1500。
測定:精密稱取奈拉濱適量,用流動相溶解並稀釋製成每1ml中含奈拉濱0.125mg的溶液,作為供試品溶液。精密量取上述供試品溶液適量,加流動相製成每1ml中含1.25μg的溶液作為對照溶液;照有關物質項下的色譜條件,取對照溶液20μl注入液相色譜儀,調節檢測器靈敏度,使主成份色譜峰的峰高約為滿量程的15%,再精密量取供試品溶液與對照溶液各20μl,分別注入液相色譜儀,分別記錄色譜峰至主成份峰保留時間的3倍。
表4 影響因素試驗結果
藥品質量的高低直接關係到千百萬勞動人民身體的健康,也關係到我國經濟建設的成效、國防的鞏固和民族的興旺發達;它已遠不是醫藥工業企業本身範圍的事,而是整個民族、世界的大事。本領域技術人員均清楚知曉,在製藥技術發達的當代,藥品安全標準被不斷提升,所製備的藥品的純度也越來越高,有效地降低雜質含量,哪怕是零點幾個百分點,也可以有效地降低不良反應的發生,因此雜質含量對藥品質量和人民用藥安全至關重要。藥品從生產到流通過程中需要貯存和運輸才能治病救人,因此,貯存和運輸過程中藥品的質量顯得尤為重要,穩定性是決定藥品質量好壞的關鍵,在藥品貯存和運輸過程中,穩定性不好,雜質變化大直接影響人民用藥安全。
從上表可以看出,採用本發明的方法製得的奈拉濱晶體化合物的單雜、總雜等含量均很低,且穩定性顯著優於現有技術的奈拉濱,有效地提升了藥品安全性及存儲的穩定性,降低了不良反應的發生。
對本發明其它實施例的奈拉濱晶體化合物也進行了上述實驗例1-4,其獲得的結果相似。
實驗例6:粉針劑影響因素試驗
製劑試驗品1:本發明製劑實施例1所製備的奈拉濱粉針劑。
製劑試驗品2:本發明製劑實施例2所製備的奈拉濱粉針劑。
製劑對照品1:採用本發明製備方法製備粉針劑,所不同的是將精氨酸換成氯化鈉。
製劑對照品2:採用本發明製備方法製備粉針劑,所不同的是採用的奈拉濱晶體為按照CN103172687A實施例1的方法製得奈拉濱晶體化合物。
製劑對照品3:採用本發明製備方法製備粉針劑,所不同的是採用的奈拉濱晶體為按照CN103172687A實施例1的方法製得奈拉濱晶體化合物,且將精氨酸換成氯化鈉。
將製劑試驗品模擬上市包裝,在高溫60℃、光照4500Lx、高溼(RH90%±5%)條件下放置10天,按穩定性重點考察項目進行檢測,與0天樣品比較,結果見表5。
表5 製劑影響因素試驗結果
由以上結果發現,本發明所製備的含有該奈拉濱晶體化合物的粉針劑純度高、雜質含量低,穩定性好。
對本發明其它製劑實施例的奈拉濱粉針劑也進行了上述試驗,其獲得的結果相似。