一種環境水體中六溴環十二烷的快速檢測方法與流程
2023-05-19 14:27:57 2
本發明涉及環境水體中有機汙染物的快速檢測分析技術,特別是涉及一種環境水體中六溴環十二烷的快速檢測方法。
背景技術:
六溴環十二烷(hbcds)是一種高溴含量的脂環族添加型阻燃劑,主要用於建築物和汽車的保溫材料、電氣和電子設備、合成樹脂及橡膠塗層等。隨著多溴聯苯醚(pbdes)在歐洲和北美地區的禁用,hbcd作為pbdes的替代品之一,使用越來越廣泛,2006年全球hbcds的產量為20000噸。在歐洲,hbcd用量僅次於四溴雙酚a(tbbpa),在我國,hbcds的生產能力約為7500噸/年,且生產量呈逐年增長趨勢。隨著hbcds的大量使用,在各環境介質中均有檢出,其中英國湖泊、比利時河流及我國太湖等水體中hbcds的濃度達到了ppb級。
六溴環十二烷的辛醇水分配較高,採用傳統的液-液萃取、索氏抽提、色譜和固相萃取法等方法,普遍存在溶劑消耗量大、處理時間長、操作步驟多、待測物易損失等特徵,並對操作人員和環境造成危害。這極大的制約了相關環境監測工作的開展,限制了對水體中六溴環十二烷殘留狀況,時空分布規律,遷移轉化規律的研究。因此,亟需建立快速、高效、環保的六溴環十二烷的檢測方法。
技術實現要素:
本發明針對水體中六溴環十二烷在分析檢測方面存在的問題,提供一種快速、簡便、所需樣品量和有機溶劑用量小的分離富集技術,實現對水體中六溴環十二烷的快速檢測技術。本發明與現有技術的區別在於,採用spme-hplc-ms/ms串聯技術富集、分離、進樣、分析檢測為一體,實現了對水體中六溴環十二烷的快速檢測。
為了實現上述目的,本發明提供了一種環境水體中六溴環十二烷的快速檢測方法,其特徵在於:該方法包括以下具體步驟:
步驟1、對待測水樣進行過濾去除水體懸浮物,準確移取10ml水樣於15ml棕色頂空樣品瓶中,加入磁力攪拌子,置於操作平臺,採用磁力加熱攪拌器攪拌加熱;
步驟2、對固相微萃取纖維頭進行老化處理,老化過程中,流動相為甲醇;流速0.5ml/min,老化處理時間30min,從而得到附著有60μmpdms/dvb塗層的固相微萃取纖維頭。
步驟3、將步驟2老化後的固相微萃取纖維頭插入頂空樣品瓶中,並浸入樣品,推出固相微萃取纖維頭,在攪拌速度600~1200r/min,溫度20~40℃條件下萃取5~40min,收回固相微萃取纖維頭;
步驟4、將固相微萃取(spme)進樣針插入與高效液相色譜-質譜/質譜(hplc-ms/ms)聯用的固相微萃取-高效液相色譜聯用(spme-hplc)接口,推出固相微萃取纖維頭,扣緊固定扣;
步驟5、待高效液相色譜(hplc)進樣針吸入甲醇樣,固相微萃取-高效液相色譜聯用儀(spme-hplc)接口的六通閥從負載(load)位置扳到噴射(inject)位置時,開始動態解吸過程,流動相通過解吸室,開始衝洗固相微萃取纖維頭,使富集的化合物洗脫下來,並隨流動相進入色譜柱和質譜進行分離和檢測;
步驟6、解吸結束,將六通閥復位至負載(load)位置,打開固定扣,回拉固相微萃取(spme)手柄推桿使固相微萃取纖維頭縮回至穿刺隔墊針中,取下固相微萃取(spme)手柄,潤洗固相微萃取纖維頭,進行下一次的萃取操作;
步驟7、繪製標準曲線,測定環境水樣中六溴環十二烷的含量。
進一步地、步驟1中,採用磁力加熱攪拌,溫度20~40℃,攪拌速度600~1200r/min。
進一步地、步驟2中,收回萃取纖維固相微萃取纖維頭做完一個樣品需依次用甲醇,超純水各洗15s。
進一步地、步驟3中,吸附處理條件為攪拌速度800r/min,溫度27℃條件下萃取10min,收回固相微萃取纖維頭。
進一步地、步驟5中,檢測條件為:
色譜柱:waterscortecstmc18,2.7um;4.6×100mm;
流動相:水、甲醇;
流速0.5ml/min;
柱溫:40℃;
質譜條件設定為:
電離方式:電噴霧離子源,負離子模式,三重四級杆質譜檢測;
噴霧電壓:-3500v;
質譜掃描方式:多反應離子監測(mrm);入口電壓ep=-10.00v,碰撞室出口電壓cxp=-15v。
進一步地,本發明還提供了環境水體中六溴環十二烷的快速檢測方法在檢測水體中六溴環十二烷方法的應用。
本方法所述環境水體中六溴環十二烷的前處理分析檢測方法與傳統方法相比較具有快速高效環保的特點。傳統方法需要的前處理時間長,操作繁瑣,且耗費大量的有機試劑,待測物易損失或汙染。
本發明的優點和積極的效果是:操作簡便、快速、高效、靈敏、環保、成本低。可用於江河、湖泊等水體中痕量物質的萃取分析,省去繁雜的實驗室前處理,大大縮短了環境水樣中六溴環十二烷的檢測時間。
附圖說明
圖1α-hbcd的標準曲線;
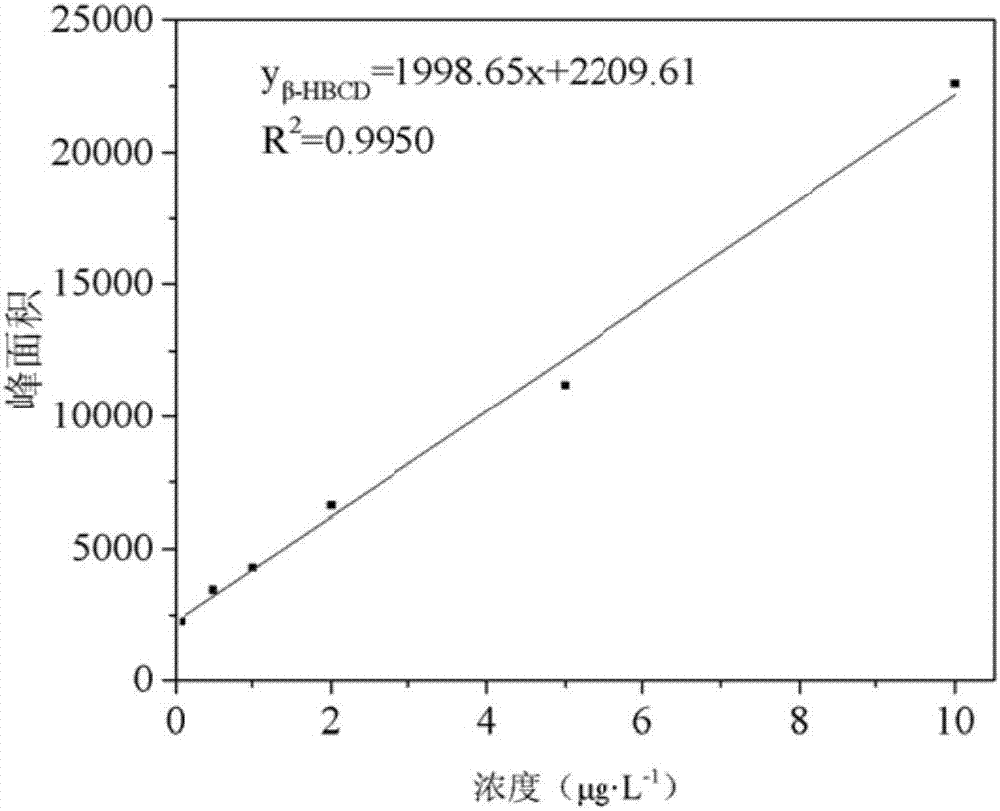
圖2β-hbcd的標準曲線;
圖3γ-hbcd的標準曲線;
具體實施方式
下面結合實施例和附圖1-3對本發明的技術方進行詳細說明。
該實施例提供了一種用於水體中六溴環十二烷的快速檢測方法,包括以下步驟:
步驟1、對待測水樣進行過濾去除水體懸浮物,準確移取10ml水樣於15ml棕色頂空樣品瓶中,加入磁力攪拌子,置於操作平臺,採用磁力加熱攪拌器攪拌加熱;
該步驟中,採用磁力加熱攪拌,溫度20~40℃,攪拌速度600~1200r/min。
步驟2、對固相微萃取纖維頭進行老化處理,老化過程中,流動相為甲醇;流速0.5ml/min,老化處理時間30min,從而得到附著有60μmpdms/dvb塗層的固相微萃取纖維頭。
該步驟中,固相微萃取纖維頭做完一個樣品需依次用甲醇,超純水各洗15s。
步驟3、將步驟2老化後的固相微萃取纖維頭插入頂空樣品瓶中,並浸入樣品,推出固相微萃取纖維頭,在攪拌速度600~1200r/min,溫度20~40℃條件下萃取5~40min,收回固相微萃取纖維頭;
優選地,該步驟中,吸附處理條件為攪拌速度800r/min,溫度27℃條件下萃取10min,收回固相微萃取纖維頭;
步驟4、將固相微萃取(spme)進樣針插入與高效液相色譜-質譜/質譜(hplc-ms/ms)聯用的固相微萃取-高效液相色譜聯用儀(spme-hplc)接口,推出固相微萃取纖維頭,扣緊固定扣;
步驟5、待高效液相色譜(hplc)進樣針吸入甲醇樣,固相微萃取-高效液相色譜聯用(spme-hplc)接口的六通閥從負載(load)位置扳到噴射(inject)位置時,開始動態解吸過程,流動相通過解吸室,開始衝洗固相微萃取纖維頭,使富集的化合物洗脫下來,並隨流動相進入色譜柱和質譜進行分離和檢測。
該步驟中,將吸附完成後的固相微萃取纖維頭直接進樣進行動態解析,利用固相微萃取高效液相色譜質譜聯用技術測定水樣中的六溴環十二烷,檢測條件為:該步驟中,將吸附完成後的固相微萃取纖維頭直接進樣進行動態解析,利用固相微萃取高效液相色譜質譜聯用技術測定水樣中的六溴環十二烷,檢測條件為:色譜柱:waterscortecstmc18,2.7um;4.6×100mm,即色譜柱規格:長100mm,內徑4.6mm,矽膠粒度2.7um;流動相:水、甲醇,見表1;流速0.5ml/min;柱溫:40℃;質譜條件設定:電離方式:電噴霧離子源,負離子模式(esi-);三重四級杆質譜檢測;噴霧電壓:-3500v;質譜掃描方式:多反應離子監測(mrm);入口電壓ep=-10.00v,碰撞室出口電壓cxp=-15v。碰撞能量見表2。
表1液相色譜梯度洗脫
表2tbbpa定性定量離子
步驟6、解吸結束,將六通閥復位至負載(load)位置,打開固定扣,回拉固相微萃取(spme)手柄推桿使固相微萃取纖維頭縮回至穿刺隔墊針中,取下固相微萃取(spme)手柄,潤洗固相微萃取纖維頭,進行下一次的萃取操作;
步驟7、繪製標準曲線,測定環境水樣中六溴環十二烷的含量。
外標法定量時可按常規方式製作標準曲線,對實際樣品進行測定,以保留時間定性,以測得目標峰面積值,代入標準曲線方程,求得樣品中六溴環十二烷的含量。
表3該實施例所述方法的加標回收率
該實施例中,在本研究優化的測定條件下,對該方法萃取頭熱穩定性、六溴環十二烷的富集倍數、線性相關係數、檢出限、加標回收率及相對標準偏差進行了考察,試驗結果表明,萃取頭在50℃依然保持良好的熱穩定性,六溴環十二烷的富集倍數超過了20倍,使用本發明所述方法對水體中六溴環十二烷的線性範圍為0.1~10ng/ml,且線性良好,線性相關係數r2為0.9901~0.9990,加標回收率為81.6%~119.6%,相對標準偏差為2.49%~11.02%,檢出限為0.0031~0.013ng/ml,以上數據說明,該方法具有較好的精密度、穩定性和重現性,可以用於水體中六溴環十二烷成分的準確測定。該實施例所述方法的加標回收率如表3所示。
雖然上面結合本發明的優選實施例對本發明的原理進行了詳細的描述,本領域技術人員應該理解,上述實施例僅僅是對本發明的示意性實現方式的解釋,並非對本發明包含範圍的限定。實施例中的細節並不構成對本發明範圍的限制,在不背離本發明的精神和範圍的情況下,任何基於本發明技術方案的等效變換、簡單替換等顯而易見的改變,均落在本發明保護範圍之內。