一種提純喜果苷的方法與流程
2023-05-04 07:35:06 2
本發明屬於天然植物單體提純技術領域,特別涉及一種提純喜果苷的方法。
背景技術:
喜樹果是我國特有樹種喜樹的果實,自古以來,喜樹在民間即為重要藥材,且可全株用藥,用於治療惡性療毒,腫瘤。喜樹果中的主要化學成分包括:喜樹鹼、10-羥基喜樹鹼、11-甲氧基喜樹鹼、脫氧喜樹鹼、喜樹次鹼、白樺酯酸及喜果苷。研究發現,喜果苷和喜樹鹼類化合物均有抗癌活性,喜果苷還具有抗菌,抗病毒,抗炎,陣痛,解熱的用途。
由喜樹果中提取喜果苷,具有重要實用價值。目前提取分離喜果苷的方法主要是柱色譜法,如:歐來良等人(CN 1295239C,2007)將喜樹果用乙醇溶液滲漉或乙醇溶液25-80℃熱提取、提取液通入裝有大孔吸附樹脂(AL-2吸附樹脂)的樹脂柱中吸附、用乙醇水溶液洗滌和解吸、流出液旋蒸濃縮和重結晶等步驟得喜果苷產品;王瑞芳等人(應用化學,2005,22(1):24-29)用合成的大孔超高交聯吸附樹脂(Rf18)及其甲胺化(Rs3)、N-甲基乙醯胺化(Rs4)、二甲胺化(Rs5)、三甲胺化(Rs6)後對喜樹鹼和喜果苷進行柱層析分離;史偉國等人(中國中藥雜誌,2008,32(21):2487-2489)用聚醯胺分離純化10-羥基喜樹鹼和喜果苷;王瑞芳等人(應用化學,2009,26(5):593-596)通過選用不同結構的納米吸附樹脂對喜樹果提取液中的喜樹鹼和喜果苷(CPT和VCS-LT)進行層析分離。現有柱色譜法提純喜果苷的工藝均為批處理工藝,洗脫劑耗量大,不能連續自動生產。
模擬移動床色譜(簡稱SMBC)是提純化合物的高效分離手段。模擬移動床色譜在製備色譜的基礎上,引進連續運行機制,能夠進行自動化和規模化的分離。
技術實現要素:
本發明的目的是針對現有技術中分離純化喜果苷方法存在的不足,提供了一種提純喜果苷的方法。該方法首先以乙醇水溶液提取喜樹果粗提物,再利用三帶模擬移動床色譜分離提純喜樹果粗提物中的喜果苷,得到喜果苷萃取液,再將萃取液進行濃縮和結晶後處理得到高純度的喜果苷。該方法可以規模化、高效地提純喜果苷,並且模擬移動床色譜分離過程實現了穩健、連續、自動運行,固定相和流動相能反覆利用,降低了提純成本。
一種提純喜果苷的方法,包括如下步驟:
(1)製備喜樹果粗提物,即:原料前處理
喜樹果原料粉粹後,用溶劑浸取1-3次,得到浸取液;將浸取液濃縮後,加乙醇或水和乙醇,使該混合液體積為所述浸取液體積的1/5~1/20,並且該混合液中乙醇的體積百分含量為60~100%,除去混合液中產生的沉澱,得濾液;再用水和乙醇稀釋濾液,得喜樹果粗提物進樣液;
所述喜樹果原料和溶劑的料液比為1g∶10mL~1g∶20mL;所述溶劑中乙醇的體積百分含量為50~100%,餘量為水;
所述喜樹果粗提物進樣液中喜果苷的濃度為0.1~3mg/mL,乙醇的體積百分含量為40~65%;
(2)用SMBC提純喜果苷
1)SMBC分離條件如下:
色譜柱:3~10根;
固定相:十八烷基矽烷鍵合矽膠ODS,10~60μm;
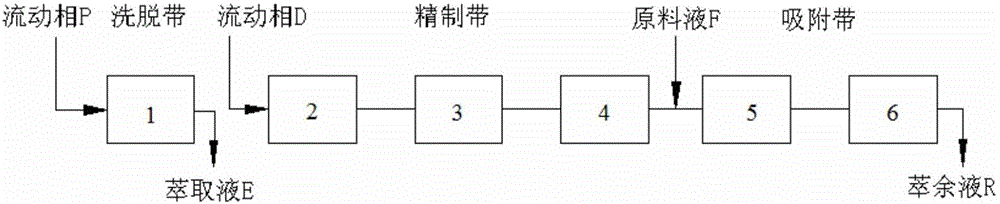
運行模式:設置三帶SMBC,三帶包括洗脫帶、精製帶和吸附帶,a為洗脫帶色譜柱數目,1≤a≤2;b為精製帶色譜柱數目,1≤b≤4;c為吸附帶色譜柱數目,1≤c≤4,運行模式用a-b-c表示;
流動相組成:洗脫帶的流動相P為乙醇與水的混合溶液或乙醇,流動相P中乙醇的體積百分含量為50~100%;精製帶和吸附帶的流動相D為乙醇與水的混合溶液,乙醇的體積百分含量為50~70%;
流動相流速:在精製帶和吸附帶中,洗脫液D流速QD為每小時1~10倍柱體積,即1~10BV/h,當洗脫液D流速QD單位為mL/min,色譜柱半徑r單位為cm,色譜柱柱長L單位為cm時,則1個BV/h相當於60QD/(πr2L);進樣液F流速QF為0.1QD~1.0QD,萃餘液R流速QR=QD+QF;在洗脫帶中,洗脫液P流速Qp為0.5QD~2QD,萃取液E流速QE=Qp;
切換時間TS:8~20min;
模擬移動床操作溫度:室溫;
2)SMBC分離過程如下:
在上述SMBC工作條件下進行SMBC分離,由原料液入口連續泵入喜樹果粗提物進樣液,SMBC自動控制系統按切換時間TS沿流動相方向將洗脫液入口、萃取液出口、原料液入口和萃餘液出口的位置同時依次移動至下一根色譜柱,從萃餘液R出口除去各種前雜質,從萃取液E出口得到喜果苷萃取液;
(3)對萃取液進行後處理
將含喜果苷萃取液濃縮至渾濁,靜置分層,棄去上層乳白色液體,用蒸餾水對下層沉澱進行清洗,直至清洗液無色;再用乙醇對下層沉澱重結晶,得到喜果苷產品。
上述的一種提純喜果苷的方法,可以用甲醇替代乙醇作為溶劑或流動相。
本發明與現有製備色譜分離技術相比,其顯著的有益效果體現在:
1、本發明中乙醇-水溶液對喜樹果原料的前處理能充分去除進樣液不溶物,保證SMBC的穩定運行,由SMBC得到的萃取液再結合後處理,能夠得到高純度的喜果苷,純度高於98%。
2、本發明利用SMBC技術能夠規模化、穩健、連續、自動、高效地從喜樹果中提純喜果苷,在SMBC的萃取液中,產品回收率高於99%,HPLC純度高於90%,固定相和流動相能反覆利用,降低了提純成本,屬於綠色環保分離過程。
附圖說明
圖1、實施例1中喜樹果進樣液的HPLC譜圖。
圖2、實施例1中的三帶SMBC體系。
圖3、實施例1中三帶SMBC萃餘液的HPLC譜圖。
圖4、實施例1中三帶SMBC萃取液的HPLC譜圖。
圖5、實施例1中經後處理的喜果苷產品的HPLC譜圖。
圖6、實施例1中經後處理的喜果苷產品的晶體掃描電鏡圖。
具體實施方式
一種提純喜果苷的方法,包括如下步驟:
(1)製備喜樹果粗提物
喜樹果原料粉粹後,用溶劑浸取1-3次,得到浸取液;將浸取液濃縮後,加乙醇或水和乙醇,使該混合液體積為所述浸取液體積的1/5~1/20,並且該混合液中乙醇的體積百分含量為60~100%,除去混合液中產生的沉澱,得濾液;再用水和乙醇稀釋濾液,得喜樹果粗提物進樣液;
所述喜樹果原料和溶劑的料液比為1g∶10mL~1g∶20mL,溶劑中乙醇的體積百分含量為50~100%,餘量為水;
所述喜樹果粗提物進樣液中喜果苷的濃度為0.1~3mg/mL,乙醇的體積百分含量為40~65%;
(2)用SMBC提純喜果苷
1)SMBC分離條件如下:
色譜柱:3~10根;
固定相:十八烷基矽烷鍵合矽膠ODS,10~60μm;
運行模式:設置三帶SMBC,三帶包括洗脫帶、精製帶和吸附帶,a為洗脫帶色譜柱數目,1≤a≤2;b為精製帶色譜柱數目,1≤b≤4;c為吸附帶色譜柱數目,1≤c≤4,運行模式用a-b-c表示;
流動相組成:洗脫帶的流動相P為乙醇與水的混合溶液或乙醇,流動相P中乙醇的體積百分含量為50~100%;精製帶和吸附帶的流動相D為乙醇與水的混合溶液,乙醇的體積百分含量為50~70%;
流動相流速:在精製帶和吸附帶中,洗脫液D流速QD為每小時1~10倍柱體積,即1~10BV/h,當洗脫液D流速QD單位為mL/min,色譜柱半徑r單位為cm,色譜柱柱長L單位為cm時,則1個BV/h相當於60QD/(πr2L);進樣液F流速QF為0.1QD~1.0QD,萃餘液R流速QR=QD+QF;在洗脫帶中,洗脫液P流速Qp為0.5QD~2QD,萃取液E流速QE=Qp;
切換時間TS:8~20min;
模擬移動床操作溫度:室溫;
2)SMBC分離過程如下:
在上述SMBC工作條件下進行SMBC分離,由原料液入口連續泵入喜樹果粗提物進樣液,SMBC自動控制系統按切換時間TS沿流動相方向將洗脫液入口、萃取液出口、原料液入口和萃餘液出口的位置同時依次移動至下一根色譜柱,從萃餘液R出口除去各種前雜質,從萃取液E出口得到喜果苷萃取液;
(3)對萃取液進行後處理
將含喜果苷萃取液濃縮至渾濁,靜置分層,棄去上層乳白色液體,用蒸餾水對下層沉澱進行清洗,直至清洗液無色;再用乙醇對下層沉澱重結晶,得到喜果苷產品。
上述的一種提純喜果苷的方法,可以用甲醇替代乙醇作為溶劑或流動相。
喜果苷的檢測方法方法為:
高效液相色譜儀:Agilent 1200;分析色譜柱:Extend-C18,5μm,150mm×4.6mm I.D.;流動相:V(甲醇)∶V(水)=60∶40;檢測波長:256nm流速:1.2mL/min;柱溫:25℃。由喜果苷標準品來標定待測液喜果苷的含量。
下面結合附圖用實施例詳細描述本發明。
實施例1
1、製備喜樹果粗提物
取100g粉碎的喜樹果原料,加入1L乙醇,在60℃下超聲浸取2次,每次浸取60min,得到浸取液;然後將浸取液濃縮至75mL,加25mLH2O,靜置分層,除去該混合液產生的沉澱,取濾液;用水-乙醇(體積比5∶5)稀釋濾液,得喜樹果粗提物的進樣液,其中,喜果苷濃度為0.56mg/mL,其色譜純度為23.79%,見圖1。
2、用SMBC從喜樹果粗提物中提純喜果苷
1)SMBC分離條件如下:
色譜柱:6根,直徑1cm,長20cm;
固定相:十八烷基矽烷鍵合矽膠ODS,20~30μm;
運行模式:1-3-2,即洗脫帶:1根色譜柱;精製帶:3根色譜柱;吸附帶:2根色譜柱,如圖2所示;
流動相組成:洗脫帶的流動相P為95%乙醇;精製帶和吸附帶的流動相D為乙醇與水的混合溶液,乙醇的體積百分含量為60%;
流動相流速:在洗脫帶中,洗脫液P流速Qp為1.5mL/min,萃取液E流速QE=Qp;在精製帶和吸附帶中,洗脫液D流速QD為1.53mL/min,進樣液F流速QF為0.29mL/min,萃餘液R流速QR=QD+QF;
切換時間:9.3min;
模擬移動床操作溫度:室溫;
2)SMBC分離過程如下:
在上述SMBC工作條件下進行SMBC分離,連續由原料液入口泵入喜樹果粗提物的進樣液,SMBC自動控制系統按切換時間TS沿流動相方向將洗脫液入口、萃取液出口、原料液入口和萃餘液出口的位置同時依次移動至下一根色譜柱,從萃餘液R出口除去各種前雜質,萃餘液HPLC譜圖如圖3所示,從萃取液E出口得到喜果苷萃取液,萃取液中喜果苷的色譜純度為91.56%,收率99.28%,萃取液HPLC譜圖如圖4所示。
3、對萃取液進行後處理
將含喜果苷萃取液濃縮至渾濁,靜置分層,棄去上層乳白色液體,用蒸餾水對下層沉澱進行清洗,直至溶液無色,用乙醇對沉澱三次重結晶,得到色譜純度為99.1%的喜果苷產品,其HPLC譜圖如圖5所示,其形貌的掃描電鏡照片見圖6。
實施例2
除下述內容外,其它條件與步驟均同實施例1;
配製喜樹果粗提物進樣液:將100g喜樹果粉碎後,用乙醇與水(體積比7∶3)的混合溶液60℃下超聲浸取二次,每次超聲浸取60min,料液比1∶15,得到浸取液;將浸取液濃幹後加乙醇與水(體積比7∶3)100mL,得到混合液,除去混合液的沉澱,取濾液;用乙醇與水(體積比5∶5)稀釋濾液,得喜樹果粗提物的進樣液,其中,喜果苷的濃度為1.14mg/mL。
從SMBC萃取液E出口得到喜果苷萃取液,喜果苷的色譜純度和收率分別為91.07%和99.34%。
對萃取液進行後處理得到色譜純度為98.80%的喜果苷產品。
實施例3
除下述內容外,其它條件與步驟均同實施例1;
配製喜樹果粗提物進樣液:將100g喜樹果粉碎後,用乙醇與水(體積比7∶3)的混合溶液超聲浸取二次,每次超聲浸取60min,料液比1∶15,得到浸取液;浸取液濃幹後加乙醇與水(體積比6∶4)150mL,得到混合液,除去混合液的沉澱,得濾液;用乙醇與水(體積比6∶4)稀釋濾液,得喜樹果粗提物的進樣液,其中,喜果苷的濃度為1.0mg/mL。
色譜柱:4根,直徑2cm,長20cm;
固定相:十八烷基矽烷鍵合矽膠ODS,30~40μm;
運行模式:1-1-2;
洗脫液P流速Qp為6mL/min,萃取液E流速QE=Qp;洗脫液D流速QD為6mL/min,進樣液F流速QF為1.2mL/min,萃餘液R流速QR=QD+QF;
從SMBC萃取液E出口,得到喜果苷萃取液,喜果苷色譜純度91.90%,回收率99.50%。
對萃取液進行後處理得到色譜純度為98.85%的喜果苷產品。