一種具有靶向功能仿病毒結構高分子囊泡及其製備與應用的製作方法
2023-04-22 17:00:57 1
本發明屬於高分子材料領域,涉及一種具有靶向功能仿病毒結構高分子囊泡及其製備方法,具體涉及一種由透明質酸接枝的兩親性嵌段共聚物與其他兩親性嵌段共聚物混合自組裝成具有仿病毒表面突觸結構的高分子囊泡及其製備方法與應用。
技術背景
惡性腫瘤是一種是危害人類生命健康的嚴重疾病,其防治仍依然是當今生命科學研究領域的一個難點。常規的化學療法是一種全身性的療法,在相關治療手段中是首選,適用於大部分實體腫瘤或血液腫瘤。然而,已知的化療藥物仍普遍存在毒性強、對腫瘤靶向識別性差的問題,在殺死腫瘤細胞的同時也對人體正常組織細胞造成很大的傷害。長期治療更會致使腫瘤細胞產生耐藥性,對人體的毒副作用很大等缺點。因此,如何選擇性地殺死腫瘤細胞而不造成機體正常組織細胞損傷成為治療的關鍵問題。選擇性或針對腫瘤細胞進行靶向治療是解決此關鍵問題的首選途徑。在自然界中,病毒是一種具有靶向性的基因載體,其主要由核酸分子和蛋白構成的外衣殼組成。病毒衣殼能屏蔽環境中的潛在破壞因素,如核酸酶、酸鹼度等,具有保護基因遺傳物質的功能,同時直接參與病毒的感染過程,實現對宿主細胞表面的特異性吸附。受病毒結構和功能的啟示,科學家們一直在不斷嘗試各種人工仿病毒結構和功能的自組裝聚集體用於藥物控釋和靶向治療的藥物載體,其中以高分子囊泡最為典型。高分子囊泡是一類由雙親性嵌段共聚物有序組合而成的中空球體,其球體的外殼膜層由雙親性分子構成,而內部親水空腔可以用於包裹藥物。高分子囊泡膜結構可分為三層:兩個內外側的親水層和一個處於內外親水層中間的疏水層,疏水層由於內外表面的親水層保持穩定。通常,高分子囊泡是由單一種類的兩親性嵌段共聚物自組裝而成,其囊泡表面呈「光滑」狀態。而本發明通過兩種及以上嵌段共聚物製備的仿病毒核衣殼及表面刺突結構的高分子囊泡是一種非常具有潛力的藥物載體。高分子囊泡可通過「特洛伊木馬」策略包載基因、藥物,並避免其中體內循環中被免疫系統辨識、吞噬。此外,通過嵌段共聚物設計與合成,如對進行功能基團接枝修飾,可使高分子囊泡表面帶有特定功能性配體從而具有對特定細胞的靶向性。
目前,同時具有針對腫瘤細胞靶向功能以及仿病毒結構高分子囊泡還未見有公開報導。
技術實現要素:
基於獲得結構與功能上仿病毒結構,同時賦予高分子囊泡靶向腫瘤細胞功能的需求,本發明的目的在於提供一種含有透明質酸接枝嵌段共聚物構建的具有腫瘤細胞靶向功能及仿病毒結構的高分子囊泡的製備方法。
本發明的另一目的在於提供由上述方法得到的具有靶向功能仿病毒結構高分子囊泡。
本發明的目的通過以下技術方案實現:
一種具有靶向功能仿病毒結構高分子囊泡的製備方法,具體包括以下步驟:
s1:將含有氨基的透明質酸(ha)與含有羧基的疏水性聚合物進行醯胺反應,得到透明質酸(ha)接枝的兩親性嵌段共聚物;所述含有羧基的疏水性聚合物在反應之前需進行羧基活化處理;
s2:將透明質酸ha接枝的兩親性嵌段共聚物與其他兩親性嵌段共聚物通過溶劑交換法或水合法製備成具有靶向功能仿病毒結構高分子囊泡。
步驟s1中所述含有氨基的透明質酸是通過二元胺與透明質酸反應得到。所述二元胺的結構為h2n-r-nh2,r為有機基團;所述二元胺優選為脂肪二元胺,更優選為丁二胺。
步驟s1中所述含有氨基的透明質酸的具體製備步驟為:
將透明質酸溶於緩衝溶液中,加入二元胺,恆溫攪拌,加入還原劑反應,反應完成後透析,凍幹,得到端氨基化透明質酸即含有氨基的透明質酸。
所述恆溫攪拌的溫度為40~60℃,優選為50℃;所述恆溫攪拌的時間為20~36h,優選為24h;
所述二元胺優選為1,4-丁二胺;所述還原劑為硼氰化鈉;
所述反應的時間為2~4天,優選為3天,所述還原劑分2~4次加入;
所述緩衝溶液優選為醋酸鹽緩衝液。
所述透明質酸ha和二元胺摩爾比為1:(1~5),還原劑與透明質酸質量比為(1~3):5。
步驟s1中所述含有羧基的疏水性聚合物在反應之前需進行羧基活化處理,活化處理具體為:在有機溶劑中,採用活化劑將含有羧基的疏水性聚合物進行羧基活化,得到羧基活化的產物;所述活化劑為1-(3-二甲氨基丙基)-3-乙基碳二亞胺鹽酸鹽(edc)與n-羥基琥珀醯亞胺(nhs)。
所述羧基活化的時間為10~36h,優選為24h。
所述含有羧基的疏水性聚合物、edc與nhs的摩爾比為(1~2.5):5:5;
所述含有羧基的疏水性聚合物進行羧基活化後需進行沉澱洗滌,乾燥處理。
所述羧基活化完成後,反應母液需進行沉澱,所述沉澱劑為甲醇/乙醚(75/25v/v)混合溶劑。
步驟s1中所述含有氨基的透明質酸與含有羧基的疏水性聚合物的摩爾比為1:1。
步驟s1中所述醯胺反應需加入n,n-二異丙基胺或n,n-二異丙基乙胺,所述含有羧基的疏水性聚合物與n,n-二異丙基胺或n,n-二異丙基乙胺的摩爾比為1:(1.5~2)。
步驟s1中所述醯胺反應以有機溶劑為反應介質,所述有機溶劑優選為二甲基亞碸(dmso)。
步驟s1中所述醯胺反應的溫度為40~60℃,優選為50℃;醯胺反應的時間為36~72h,優選為48h。
步驟s1中所述醯胺反應完成後,反應液需透析,凍幹處理,得到最終產物。
步驟s1中所述含有羧基的疏水性聚合物為端基為羧基的聚己內酯(pcl)、端基為羧基的聚乳酸(pla)、端基為羧基的聚乙烯吡咯烷酮(pvp)中的一種;
步驟s2中所述其他兩親性嵌段共聚物為聚乙二醇-b-聚己內酯(peg-b-pcl)、聚乙二醇-b-聚乳酸(peg-b-pla)、聚乙二醇-b-聚乳酸-羥基乙酸共聚物(peg-b-plga)中的一種以上。
步驟s2中所述透明質酸ha接枝的兩親性嵌段共聚物與其他兩親性嵌段共聚物的質量比為1:(5~20)。
步驟s2中所述溶劑交換法為:將透明質酸ha接枝的兩親性嵌段共聚物與其他兩親性嵌段共聚物溶於有機溶劑中,加入水或磷酸鹽緩衝液(pbs),持續攪拌使有機溶劑完全揮發,得到高分子囊泡溶液;
步驟s2中所述水合法為:將透明質酸ha接枝的兩親性嵌段共聚物與其他兩親性嵌段共聚物溶於有機溶劑中,完全溶解後,成膜;將膜與水或pbs緩衝液進行攪拌,得到高分子囊泡溶液;攪拌的時間為2~5天;所述成膜為真空乾燥成膜;
所述溶劑法中有機溶劑為四氫呋喃(thf)、n,n-二甲基甲醯胺(dmf)、n,n-二甲基乙醯胺(dmac)、二氯甲烷(dcm)、氯仿(chcl3)中的一種以上。
所述水合法中有機溶劑為四氫呋喃(thf)、n,n-二甲基甲醯胺(dmf)、n,n-二甲基乙醯胺(dmac)、二氯甲烷(dcm)、氯仿(chcl3)中的一種以上。
步驟s2中所述溶劑法或水合法製備具有靶向功能仿病毒結構高分子囊泡時,所得高分子囊泡溶液需進行純化分離。
所述純化分離的方法為透析法或液相色譜柱法中的一種以上。
所述液相色譜柱法進行分離時,將高分子囊泡溶液從色譜柱上方加入,之後加入pbs緩衝液使高分子囊泡溶液按照尺寸大小在不同的淋出體積/保留時間從色譜柱下方分離出,按照所需尺寸收集。
步驟s2中所述具有靶向功能仿病毒結構高分子囊泡的尺寸為50nm~400nm。
步驟s2中所述具有靶向功能仿病毒結構高分子囊泡以溶液形式存在,其濃度為5mg/ml~30mg/ml,優選為10mg/ml~20mg/ml。
所述具有靶向功能仿病毒結構高分子囊泡通過上述方法得到。
所述具有靶向功能仿病毒結構高分子囊泡用於藥物載體和/或製備治療腫瘤藥物。
本發明的透明質酸(ha)是一種d-葡萄糖醛酸和n-乙醯-d-葡萄胺兩種單糖重複排列的直線性多糖。ha基團能與腫瘤細胞中受體cd44配對,這種受體與ha相互作用,可調節腫瘤發展和轉移過程中的細胞行為。基於ha能與腫瘤細胞表面cd44特異性受體結合的特性,給予藥物載體輸送藥物的靶向性,引導藥物載體被腫瘤細胞內吞,從而提高藥物利用率,增強藥物療效。
與現有技術相比,本發明具有如下優點以及有益效果:
本發明採用具有透明質酸基團的嵌段共聚物與其他嵌段共聚物共混,通過水合法或溶劑法自組裝製備高分子囊泡;通過透明質酸基團的嵌段共聚物與其他嵌段共聚物的選用以及兩嵌段共聚共混比例的調控使得高分子囊泡具有仿病毒結構,而嵌段共聚物中由於透明質酸基團的存在,使得高分子囊泡具有腫瘤細胞靶向性。總之,本發明的方法重複性好,所獲得高分子囊泡具有仿病毒結構,同時其靶向性好,具有較好的應用前景。本發明的方法易於獲得不同形貌和功能性的高分子囊泡,並應用於藥物釋放等多種領域。
附圖說明
圖1為實施例1製備的ha接枝的兩親性嵌段共聚物的1h-nmr核磁氫譜圖
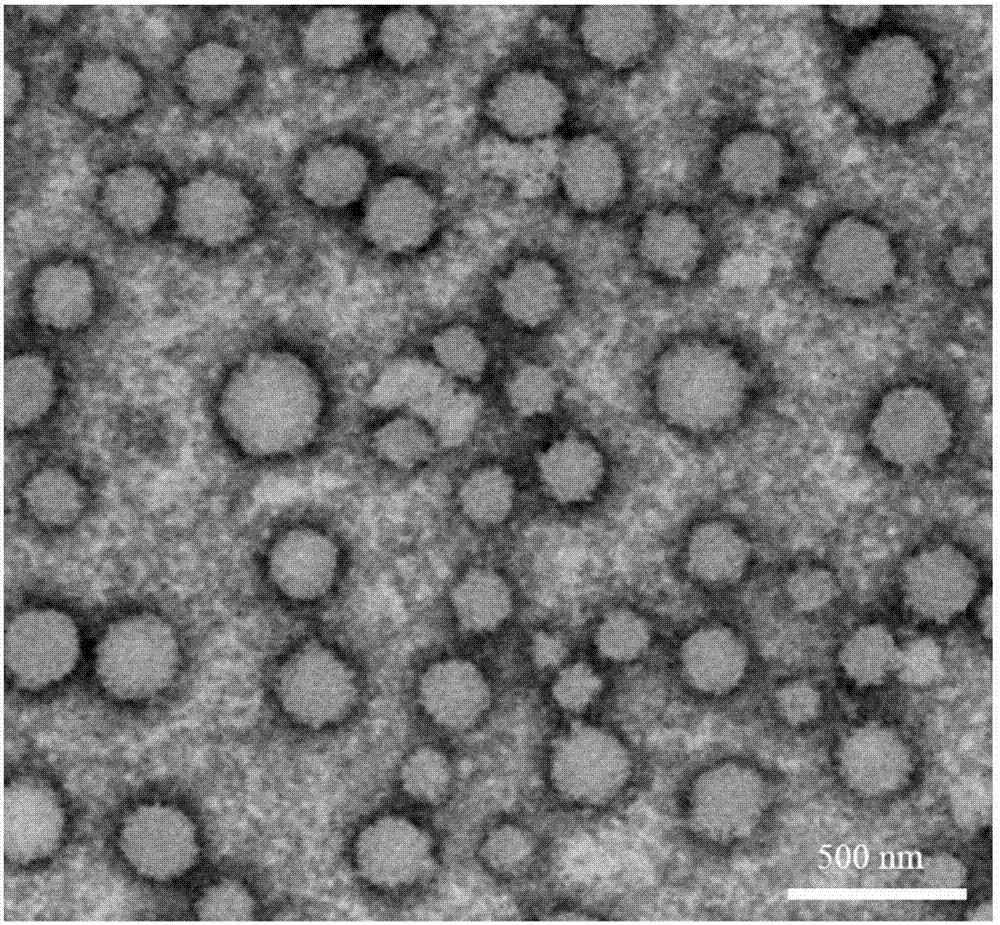
圖2為實施例1製備的高分子囊泡的透射電子顯微鏡圖(tem),放大倍數為2萬倍。
具體實施方式
下面結合實施例和附圖對本發明作進一步解釋說明,但本發明的實施方式不限於此。
實施例1
(1)將1.0g透明質酸(mw=5000,0.2mmol)溶於30ml的醋酸鹽緩衝液(ph=5.6,醋酸/醋酸鈉)中,完全溶解後,加入1.0ml的1,4-丁二胺(0.4mmol),50℃恆溫條件下,磁力攪拌24小時;之後在保持溫度攪拌條件不變的情況下,每天加入0.2g的硼氰化鈉,共三次,採用透析袋在去離子水中透析1天,將袋中物-20℃條件下冷凍乾燥得到端氨基化透明質酸;
(2)稱取1.0g的pcl-cooh(端羧基的pcl,mw=10,000,0.1mmol),溶於10ml的甲醇中,加入0.2mmol的edc與0.2mmol的nhs,室溫下攪拌24h,使用10ml甲醇/乙醚(75/25v/v)沉澱產物,70℃真空乾燥3h,得到nhs/羧基活潑中間酯即羧基活化的產物;
(3)稱取總質量為0.5g的端氨基化透明質酸和nhs/羧基活潑中間酯(摩爾比為1:1)溶於20mldmso中,加入20μl的n,n-二異丙基胺(0.05mmol),在50℃的條件下攪拌反應48小時,採用透析袋在去離子水中透析1天,將袋中物在-20℃條件下冷凍乾燥,得到ha接枝的兩親性嵌段共聚物,其1h-nmr氫譜見圖1;
(4)稱取合成的ha接枝的兩親性嵌段共聚物和peg45-b-pla140混合物,總質量為25mg,質量比為1:10,完全溶解在2.5ml二氯甲烷中,加入5ml去離子水,充分攪拌使有機溶劑揮發完全,後移入透析袋中,透析3天,得到平均直徑200nm的10mg/ml高分子囊泡溶液。所述高分子囊泡的透射電鏡圖如圖2所示,其中可見囊泡的直徑約200nm。
實施例2
(1)將2.0g透明質酸(mw=5,000,0.4mmol)溶於30ml醋酸鹽緩衝液(ph=5.6,醋酸/醋酸鈉)中,完全溶解後,加入1.0ml的1,4-丁二胺(0.4mmol),50℃恆溫條件下,磁力攪拌24小時;之後在保持溫度攪拌條件不變的情況下,每天加入0.2g的硼氰化鈉,共三次,採用透析袋在去離子水中透析1天,將袋中物-20℃條件下冷凍乾燥,得到端氨基化透明質酸;
(2)稱取1.0g的pcl-cooh(端羧基的pcl,mw=10,000,0.1mmol),溶於10ml的甲醇中,加入0.2mmol的edc與0.2mmol的nhs,室溫下攪拌24h,使用10ml甲醇/乙醚(75/25v/v)沉澱產物,70℃真空乾燥3h,得到nhs/羧基活潑中間酯;
(3)稱取總質量為0.5g的端氨基化透明質酸和nhs/羧基活潑中間酯(摩爾比為1:1)溶於20mldmso中,加入20μl的n,n-二異丙基胺(0.05mmol),在溫度50℃的條件下攪拌反應48小時,透析1天處理後,-20℃條件下冷凍乾燥得到ha接枝的兩親性嵌段共聚物;
(4)稱取合成的ha接枝的兩親性嵌段共聚物和peg45-b-pcl140總質量為25mg,質量比為1:15,完全溶解在2.5ml的thf中,向高分子溶液中加入5ml去離子水,充分攪拌後移入透析袋中,透析1天,得到10mg/ml平均直徑150nm的高分子囊泡溶液。
實施例3
(1)將1.0g透明質酸(mw=5000,0.2mmol)溶於30ml醋酸鹽緩衝液(ph=5.6,醋酸/醋酸鈉)中,完全溶解後,加入1.0ml的1,4-丁二胺(0.4mmol),50℃恆溫條件下,磁力攪拌24小時;之後在保持溫度攪拌條件不變的情況下,每天加入0.2g的硼氰化鈉,共三次,採用透析袋在去離子水中透析1天,將袋中物在-20℃條件下冷凍乾燥,得到端氨基化透明質酸;
(2)稱取2.0g的pvp-cooh(端羧基的pvp,mw=10,000,0.2mmol),溶於10ml的甲醇中,加入0.4mmol的edc與0.4mmol的nhs,室溫下攪拌24h,使用10ml甲醇/乙醚(75/25v/v)沉澱產物,70℃真空乾燥3h,得到nhs/羧基活潑中間酯;
(3)稱取總質量為0.5g的端氨基化透明質酸和nhs/羧基活潑中間酯(摩爾比為1:1)溶於20mldmso中,加入20μl的n,n-二異丙基胺(0.05mmol),在溫度50℃的條件下攪拌反應48小時,透析1天處理後,-20℃條件下冷凍乾燥得到ha接枝的兩親性嵌段共聚物;
(4)稱取合成的ha接枝的兩親性嵌段共聚物和peg45-b-pla140總質量為25mg,質量比為1:15,完全溶解在2.5ml的thf中完全溶解,後經真空中除盡有機溶劑成膜,之後添加去離子水,進行磁力攪拌5天,通過液相色譜柱法純化分離,得到5mg/ml平均直徑250nm的高分子囊泡溶液。
實施例4
(1)將2.0g透明質酸(mw=5000,0.4mmol)溶於30ml醋酸鹽緩衝液(ph=5.6,醋酸/醋酸鈉)中,完全溶解後,加入1.0ml的1,4-丁二胺(0.4mmol),50℃恆溫條件下,磁力攪拌24小時;之後在保持溫度攪拌條件不變的情況下,每天加入0.1g的硼氰化鈉,共三次,採用透析袋在去離子水中透析1天,將袋中物-20℃條件下冷凍乾燥,得到端氨基化透明質酸;
(2)稱取2.0g的pla-cooh(端羧基的pla,mw=10,000,0.2mmol),溶於10ml的甲醇中,加入0.4mmol的edc與0.4mmol的nhs,室溫下攪拌24h,使用10ml甲醇/乙醚(75/25v/v)沉澱產物,70℃真空乾燥3h,得到nhs/羧基活潑中間酯;
(3)稱取總質量為0.5g的端氨基化透明質酸和nhs/羧基活潑中間酯(摩爾比為1:1)溶於20mldmso中,加入20μl的n,n-二異丙基胺(0.05mmol),在溫度50℃的條件下攪拌反應48小時,透析1天處理後,-20℃條件下冷凍乾燥得到ha接枝的兩親性嵌段共聚物;
(4)稱取合成的ha接枝的兩親性嵌段共聚物和peg45-b-pla140總質量為25mg,質量比為1:15,完全溶解在2.5ml的thf中完全溶解,後經真空中除盡有機溶劑成膜,之後添加去離子水,進行磁力攪拌2天,通過液相色譜柱法純化分離,得到5mg/ml平均直徑200nm的高分子囊泡溶液。
實施例5
(1)將1.0g透明質酸(mw=5000,0.2mmol)溶於30ml醋酸鹽緩衝液(ph=5.6,醋酸/醋酸鈉)中,完全溶解後,加入1.0ml的1,4-丁二胺(0.4mmol),50℃恆溫條件下,磁力攪拌24小時;之後在保持溫度攪拌條件不變的情況下,每天加入0.1g的硼氰化鈉,共三次,採用透析袋在去離子水中透析1天,將袋中物-20℃條件下冷凍乾燥,得到端氨基化透明質酸;
(2)稱取2.0g的pcl-cooh(端羧基的pcl,mw=10,000,0.2mmol),溶於10ml的甲醇中,加入0.2mmol的edc與0.2mmol的nhs,室溫下攪拌24h,使用10ml甲醇/乙醚(75/25v/v)沉澱產物,70℃真空乾燥3h,得到得到nhs/羧基活潑中間酯;
(3)稱取總質量0.5g稱取總質量為0.5g的端氨基化透明質酸和nhs/羧基活潑中間酯(摩爾比為1:2)溶於20mldmso中,加入20μl的n,n二異丙基胺(0.05mmol),在溫度50℃的條件下攪拌反應48小時,透析1天處理後,-20℃條件下經冷凍乾燥得到ha接枝的兩親性嵌段共聚物;
(4)稱取ha接枝的兩親性嵌段共聚物和peg45-b-plga100總質量為30mg,質量比為1:20,完全溶解在3ml的氯仿(chcl3)中,真空乾燥箱中乾燥除去有機溶劑,加入去離子水,磁力攪拌5天,透析3天,得到平均直徑300nm的30mg/ml高分子囊泡溶液。
實施例6
(1)將2.0g透明質酸(mw=5,000,0.4mmol)溶於30ml配製而成的醋酸鹽緩衝液(ph=5.6,醋酸/醋酸鈉)中,完全溶解後,加入1.0ml的1,4-丁二胺(0.4mmol),50℃恆溫條件下,磁力攪拌24小時;之後在保持溫度攪拌條件不變的情況下,每天加入0.1g的硼氰化鈉,共三次,採用透析袋在去離子水中透析1天,將袋中物-20℃條件下冷凍乾燥,得到端氨基化透明質酸;
(2)稱取1.5g的pvp-cooh(端羧基的pvp,mw=10,000,0.15mmol),溶於10ml的甲醇中,加入0.8mmol的edc與0.8mmol的nhs,室溫下攪拌24h,使用10ml甲醇/乙醚(75/25v/v)沉澱產物,70℃真空乾燥3h,得到nhs/羧基活潑中間酯;
(3)稱取總質量為0.5g的端氨基化透明質酸和nhs/羧基活潑中間酯(摩爾比為1:1)溶於20mldmso中,加入20μl的n,n-二異丙基胺(0.04mmol),在溫度50℃的條件下攪拌反應48小時,透析1天處理後,-20℃條件下冷凍乾燥得到ha接枝的兩親性嵌段共聚物;
(4)稱取合成的ha接枝的兩親性嵌段共聚物和peg45-b-pcl120質量為30mg,質量比為1:5,完全溶解在5ml的n,n-二甲基甲醯胺(dmf)中完全溶解,後經真空中除盡有機溶劑成膜,之後添加去磷酸鹽緩衝液(pbs),進行磁力攪拌3天,通過液相色譜柱法純化分離,得到5mg/ml平均直徑50nm的高分子囊泡溶液。