一種新型小分子結構及其在檢測藍綠藻肝毒素方面的應用的製作方法
2023-05-12 02:21:06 2
本發明屬於食品安全
技術領域:
。更具體地,涉及一種新型小分子結構及其在檢測藍綠藻肝毒素方面的應用。
背景技術:
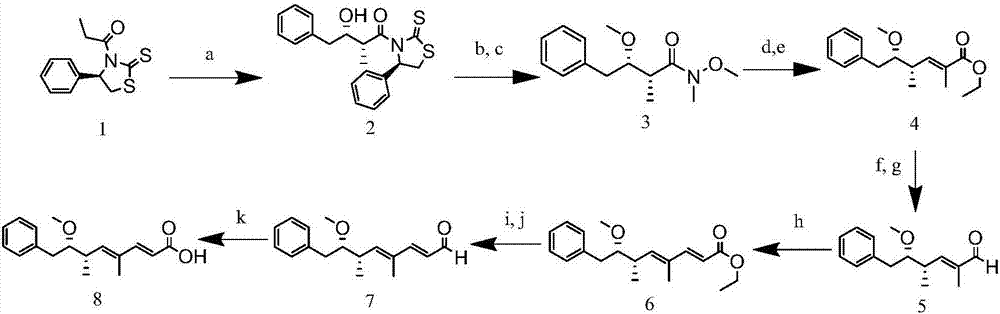
:隨著工業化、城市化的發展,大量營養物注入水中造成水體富營養化,而致使藻類迅猛生長,藻類代謝產生包括藍綠藻肝毒素(微囊藻毒素,節球藻毒素)在內的有毒物質,危害著人類健康。我國是世界上藻災害最為嚴重的國家之一,淡水湖泊中發生的水華80%是有毒的,而微囊藻毒素分布最廣、毒性最大。微囊藻毒素(microcystins,mcs)主要由淡水藻類銅綠微囊藻產生。mcs是一類單環七肽肝毒素,由於多肽中兩種可變胺基酸組成的不同,主要有l、r、y、w和f,分別代表亮氨酸、精氨酸、酪氨酸、色氨酸和苯丙氨酸,因而具有多種異構體,其中存在最普遍,含量最多的是mc-lr、mc-yr和mc-rr三種,其他還包含mc-wr、mc-la、mc-lw、mc-ly和mc-lf等在內的80多種。而adda基團(3-氨基-9-甲氧基-2,6,8-三甲基-10-苯基-4,6-二烯酸)是一種特殊的β-胺基酸,是微囊藻毒素類物質共同擁有的結構,且其為主要表達毒性的部分。mcs分布最廣泛,它對蛋白磷酸酶1和蛋白磷酸酶2a具有抑制作用,為腫瘤促進劑。1998年世界衛生組織(who)的《用水衛生標準》中建議飲用水中mcs標準為1.0μg/l。我國生活飲用水衛生標準(gb5749-2006)的頒布,將飲用水中微囊藻毒素含量限制為1μg/l,該標準的實施對水源水的質量提出了更高的要求。節球藻毒素(nodularin,nod)也是一種藍藻水華腐爛後釋放於水中的毒素之一,主要存在於泡沫節球藻中。nod為環狀五肽肝毒素,已分離得到7種節球藻毒素異構體,也都具備adda基團,是絲氨酸/蘇氨酸蛋白磷酸酶1(pp1)和蛋白磷酸酶2a(pp2a)的抑制劑,同時也是腫瘤誘發擠和促進劑,其對人類的危害可見一斑,但目前仍沒有nod的限量標準,但設置其限量標準勢在必行。目前,微囊藻毒素和節球藻毒素的檢測方法主要有薄層色譜(thinlayerchromatography,tlc)、高效液相色譜(highperformanceliquidchromatography,hplc)、氣相色譜(gaschromatography,gc)、質譜(masssepectrum,ms)以及高效液相-質譜聯用(highperformanceliquidchromatography-masssepectrum,hplc-ms)等方法。這些方法雖然準確度高,但其儀器設備昂貴、樣品前處理複雜繁瑣、檢測成本高且需要專業人員操作。而免疫層析檢測方法具備快速、靈敏、準確、操作簡便等特點,且對樣品純度要求不高,特別適用於大批量樣品的檢測。目前已報導的藍綠藻肝毒素(多種微囊藻毒素類似物和節球藻毒素)製備的抗體大多使用mc-lr和nod偶聯蛋白製備抗原、抗體,而其抗體特性也有很大差異,廣譜性也受到免疫原和包被原的限制。技術實現要素:本發明要解決的技術問題是克服現有技術中缺乏同時檢測多種微囊藻毒素結構類似物和節球藻毒素的免疫學檢測方法,提供一種近似於共同基團adda的新型小分子物質,以增加檢測方法的廣譜識別性能,製備出靈敏性度高且廣譜性強的半定量藍綠藻肝毒素免疫層析試紙條,適用於多種微囊藻毒素類似物和節球藻毒素,且具有快速、直觀、易於操作等特點,可作為檢測主要藍綠藻肝毒素的有效手段,具有廣闊的發展前景。本發明的目的是提供一種新型小分子免疫原結構。本發明另一目的是提供所述小分子免疫原結構在製備藍綠藻肝毒素廣譜免疫層析試紙條中的應用。本發明上述目的通過以下技術方案實現:一種新型小分子結構lh-1,其結構式如式(i)所示:該新型小分子結構lh-1採用系統命名法命名為:(2e,4e,6s,7s)-7-甲氧基-4,6-二甲基-8-苯辛基-2,4-二烯酸(即:(2e,4e,6s,7s)-7-methoxy-4,6-dimethyl-8-phenylocta-2,4-dienoicacid)。本發明首先獲得了一種特殊的新型小分子結構作為免疫原,將該小分子及微囊藻毒素mc-lr分別與載體蛋白偶聯製得人工抗原,利用將該小分子人工抗原乳化後免疫兔,分離純化兔血清得到免疫製備多克隆抗體,製備抗體標記的納米金顆粒,將mc-lr人工抗原包被於硝酸纖維膜上作為檢測帶,羊抗兔二抗作為控制帶,依據免疫競爭法原理建立得快速檢測藍綠藻肝毒素的廣譜性免疫層析試紙條。該免疫層析試紙條靈敏性度高、廣譜性強,可半定量檢測藍綠藻肝毒素,適用於多種微囊藻毒素類似物和節球藻毒素,且具有快速、直觀、易於操作等優勢。所述新型小分子結構lh-1的製備方法是使用n-丙醯基硫偶氮烷硫酮經過縮二脲醛醇縮合、反式醯胺化反應、烷基化反應、還原反應、wittig反應、還原反應、氧化反應、wittig反應、還原反應、氧化反應、氧化反應共11步反應生成新型小分子。具體地,新型小分子結構的製備方法包括以下步驟:將右旋n-丙醯基硫偶氮烷硫酮溶於無水二氯甲烷中,冷卻後依次加入四氯化鈦、n,n-二異丙基乙胺、n-甲基吡咯烷酮和苯乙醛,經縮二脲醛醇縮合純化後生成黃色固體狀的醛醇2。將醛醇2溶於無水二氯甲烷,依次加入n,o-二甲基羥胺鹽酸鹽和咪唑,經反式醯胺化反應,純化後得黃色油狀物中間體weinreb醯胺。將碘甲烷溶於無水四氫呋喃中,加入固體碳酸鈉攪拌,將混合物冷卻,脫脂棉過濾並加入到先前獲得的weinreb醯胺中,之後加入氫化鈉,純化後得到黃色油狀物3。將weinreb醯胺3溶於無水二氯甲烷中,冷卻後滴加二異丁基氫化鋁的無水甲苯溶液,純化後過濾,並真空濃縮得到橙色油狀中間體醛。將新製備的醛溶於無水甲苯中,加入穩定的磷葉立德(1-乙氧基羰基亞乙基)三苯基正膦,回流攪拌過夜,wittig反應純化後得到黃色油狀烯烴4。將烯烴4溶於無水四氫呋喃中,冷卻後滴加1.0m二異丁基氫化鋁的無水甲苯溶液攪拌,還原反應純化後得淡黃色油狀的烯丙醇。將烯丙醇溶於無水甲苯中,加入活化的二氧化錳,將所得懸浮液在55℃劇烈攪拌2h,氧化反應純化後得到黃色油狀物醛5。將新製備的醛5溶於無水甲苯中,加入磷葉立德(1-乙氧基羰基亞甲基)三苯基正膦,wittig反應純化後得到黃色油狀物酯6。將酯6溶於無水四氫呋喃中,冷卻後滴加二異丁基氫化鋁的無水甲苯溶液,還原反應純化後得到黃色油狀中間體醇。將醇溶於無水甲苯中,冷卻加入二氧化錳,氧化反應純化後減壓濃縮濾液得到黃色油狀醛7。將醛7溶於二甲基亞碸中,加入1,3,5-三甲氧基苯,次氯酸鈉和磷酸二氫鈉,經純化即得新型小分子結構。更優選地,所述新型小分子結構物質lh-1的製備方法,包括以下步驟:(a)縮二脲醛醇縮合:室溫下,稱取26.2g右旋n-丙醯基硫偶氮烷硫酮(cas:1217320-19-8)溶於250ml無水二氯甲烷中攪拌,之後在丙酮中用乾冰冷卻至-78℃,逐滴向上述溶液中加入12ml四氯化鈦,所得混合物攪拌15min形成鈦絡合物,再向上述溶液中加入18.9ml蒸餾過的n,n-二異丙基乙胺,將混合物接著攪拌40min形成相應的烯醇化鈦,再加入20.0ml新蒸餾的n-甲基吡咯烷酮,10min後,加入13.4ml新蒸餾的苯乙醛,將上述溶液持續在-78℃再放置30min後溫熱至4℃,並在該溫度下再攪拌1h。通過tlc監測反應,展開劑為環己烷-乙酸乙酯(7:3,v/v),最後加入300ml50%nh4cl水溶液猝滅反應。將有機層用300ml1.0n的hcl和300ml10%na2co3和300ml鹽水洗滌兩次,再用na2so4乾燥。得到黃色固體狀的醛醇2。(b)反式醯胺化反應:將37.1g醛醇2溶於無水150ml二氯甲烷中,依次加入14.6gn,o-二甲基羥胺鹽酸鹽和27.2g咪唑,將此混合物在室溫下攪拌12h。通過tlc監測反應,展開劑為環己烷-乙酸乙酯(7:3,v/v)。然後用300ml去離子水將有機層洗滌3次,再用na2so4乾燥,過濾並真空濃縮。所得殘餘物通過矽膠柱層析純化,稱取600g矽膠填柱,以環己烷和乙酸乙酯(acoet,30-50%)作為流動相梯度洗脫,得到黃色油狀物中間體weinreb醯胺。(c)烷基化反應:將40.1ml碘甲烷溶於140ml無水四氫呋喃中,再將此溶液與1.38g固體na2co3混合攪拌10min後冷卻至4℃,通過脫脂棉過濾並加入到先前獲得的15.1gweinreb醯胺中,之後,在4℃下向上述溶液中加入2.49g的nah(60%分散在礦物油中),將反應混合物在室溫下攪拌4h。通過tlc監測反應,展開劑為環己烷-乙酸乙酯(6:4,v/v)。再加入1.24g的nah(60%油溶液),將反應混合物在室溫下再攪拌2h。通過tlc監測反應,展開劑為環己烷-乙酸乙酯(6:4,v/v)。冷卻至4℃後,過量的nah通過加入150ml飽和nh4cl去除。混合物用acoet萃取兩次,有機層再用250ml鹽水洗滌,之後用na2so4乾燥,過濾並減壓濃縮。用300g矽膠填柱後,用環己烷-acoet(6:4,v/v)作為流動相純化,得到黃色油狀weinreb醯胺3。(d)還原反應:將10.71gweinreb醯胺3溶於100ml無水二氯甲烷中,將所得溶液用在丙酮中的乾冰冷卻至-78℃。在80min內滴加100ml的1.0m二異丁基氫化鋁的無水甲苯溶液。通過tlc監測反應,展開劑為環己烷-乙酸乙酯(7:3,v/v),再依次加入40ml甲醇和40ml飽和nh4cl猝滅反應,之後將該溶液升溫至室溫攪拌30min。然後將混合物通過545墊,並用300ml苯乙基洗滌,合併有機相後用200ml2.0nhcl洗滌,再用mgso4乾燥,過濾並真空濃縮得到橙色油狀中間體醛。(e)wittig反應:將8.48g新製備的醛溶於275ml無水甲苯中,加入18.52g磷葉立德(1-乙氧基羰基亞乙基)三苯基正膦,將所得混合物回流反應12h。通過tlc監測反應,展開劑為環己烷-乙酸乙酯(8:2,v/v)。將反應混合物減壓濃縮,所得殘餘物通過矽膠柱純化,用0-10%acoet的環己烷溶液作為流動相,得到黃色油狀烯烴4。(f)還原反應:將7.59g烯烴4溶於55ml無水四氫呋喃中,並將所得溶液用丙酮中的乾冰冷卻至-78℃,在1h內向上述溶液滴加68ml1.0m二異丁基氫化鋁的無水甲苯溶液,在該溫度下攪拌90min,並通過tlc監測反應,展開劑為環己烷-乙酸乙酯(8:2,v/v),之後依次加入30ml甲醇和30mlnh4cl淬滅反應。使所得溶液升溫至室溫30min後將混合物用545墊過濾並用150ml二氯甲烷洗滌,再將有機層用200ml1.0nhcl洗滌,合併有機層並用mgso4乾燥,過濾並真空濃縮,再經過矽膠柱層析純化,以0-20%acoet的環己烷溶液作為流動相梯度洗脫,得到淡黃色油狀的烯丙醇。(g)氧化反應:將3.4g烯丙醇溶於100ml無水甲苯中,將所得溶液冷卻至0℃,再加入13.6gmno2,將所得懸浮液在55℃劇烈攪拌2h。通過tlc監測反應,展開劑為環己烷-乙酸乙酯(8:2,v/v),將所得溶液用545墊過濾,並用大量二氯甲烷洗滌。將所得濾液減壓濃縮,得到黃色油狀物醛5。(h)wittig反應:將3.39g新製備的醛5溶於80ml無水甲苯中,加入6.06g磷葉立德(1-乙氧基羰基亞甲基)三苯基正膦,將所得反應混合物攪拌並回流過夜。通過tlc監測反應,展開劑為環己烷-乙酸乙酯(8:2,v/v)。此後,將反應混合物用545墊過濾,用二氯甲烷洗滌並減壓濃縮。所得油狀殘餘物通過矽膠柱純化,以0-20%acoet的環己烷溶液作為流動相梯度洗脫,得到黃色油狀物酯6。(i)還原反應:將1.0g酯6溶於12ml無水四氫呋喃中,並將所得溶液用乾冰的丙酮冷卻至-78℃,在15min內向上述溶液中逐滴滴加12ml1.0m二異丁基氫化鋁的無水甲苯溶液,在-78℃下攪拌90min,並通過tlc監測反應,展開劑為環己烷-乙酸乙酯(8:2,v/v),之後在-78℃下,緩慢加入5ml甲醇和5mlnh4cl淬滅反應,然後將溶液溫熱至室溫攪拌30min。加入50ml1.0nhcl後,用acoet萃取有機相,並用mgso4乾燥,過濾並在真空下濃縮。所得殘餘物通過矽膠柱純化,以0-20%acoet的環己烷溶液作為流動相梯度洗脫,得到黃色油狀中間體醇。(j)氧化反應:將上一步所得醇全部溶於35ml無水甲苯中,冷卻至4℃。加入4.0gmno2,在50℃下攪拌1h。通過tlc監測反應,展開劑為環己烷-乙酸乙酯(7:3,v/v),並將懸浮液用545墊過濾,並用大量二氯甲烷洗滌,減壓濃縮濾液得到黃色油狀醛7。(k)氧化反應:將醛7溶於10mldmso中,再加入3.0g1,3,5-三甲氧基苯後攪拌5min,之後將2.0gnaclo2和3.0gnah2po4溶於水中,再將該水溶液逐滴加入到dmso溶液中並持續攪拌,通過tlc監測反應,展開劑為環己烷-乙酸乙酯(1:1,v/v),反應4h後使用na2s2o3猝滅反應並加入5ml1.0nhcl酸化反應,通過acoet萃取有機相併用水洗滌,再用mgso4乾燥,所得殘餘物通過矽膠柱純化,以40%acoet的正己烷溶液(0.1%hoac)作為流動相梯度洗脫,減壓濃縮濾液得到黃色油狀羧酸8,經tlc純化反應,展開劑為環己烷-乙酸乙酯(1:1,v/v)。另外,上述新型小分子結構lh-1在製備藍綠藻肝毒素檢測用人工抗原、在製備藍綠藻肝毒素檢測用抗體或在製備藍綠藻肝毒素檢測產品方面的應用,均應在本發明的保護範圍之內。一種由上述新型小分子結構lh-1與載體蛋白偶聯製得小分子人工抗原,以及由該人工抗原免疫動物獲得的抗體,也均在本發明的保護範圍之內。優選地,所述載體蛋白為鑰孔血藍蛋白(klh)等。具體是將該小分子人工抗原乳化後免疫兔,分離純化兔血清得到多克隆抗體。作為優選的實施方案,新型小分子結構多克隆抗體的製備方法包括以下步驟:用新型小分子結構偶聯鑰孔血藍蛋白(klh)的人工抗原免疫紐西蘭大白兔,分別將1mg/ml的人工抗原溶液與等量佐劑混合乳化,初次免疫與弗氏完全佐劑混合乳化後免疫,之後的加強免疫使用免疫原與弗氏不完全佐劑乳化後免疫,共免疫四次,每次間隔3周,每次免疫劑量為0.6ml/只;第四次免疫後第十天採血,採用辛酸-硫酸銨法純化抗體。另外,上述小分子人工抗原或由其製備的藍綠藻肝毒素檢測用抗體在製備藍綠藻肝毒素檢測產品方面的應用,也都在本發明的保護範圍之內。一種藍綠藻肝毒素廣譜免疫層析試紙條,是將上述抗體進行金納米顆粒標記製得抗體標記的納米金顆粒,將微囊藻毒素mc-lr人工抗原(微囊藻毒素mc-lr與載體蛋白偶聯製得)包被於硝酸纖維膜上作為檢測帶,羊抗兔二抗作為控制帶,依據免疫競爭法原理建立快速檢測藍綠藻肝毒素的廣譜性免疫層析試紙條。具體地,所述藍綠藻肝毒素廣譜免疫層析試紙條依次包括點樣孔、檢測帶和控制帶;點樣孔上噴塗新型小分子結構抗體標記的膠體金複合物,檢測帶為微囊藻毒素mc-lr的蛋白載體的偶聯物,控制帶上噴塗羊抗兔二抗;試紙的基質為硝酸纖維膜。更具體地,所述藍綠藻肝毒素廣譜免疫層析試紙條由如下方法製備得到:將新型小分子結構抗體標記的膠體金複合物噴塗金標墊(噴塗量為50~70μl/cm2,優選60μl/cm2),用7~10mg/ml(優選8mg/ml)的mc-lr-ova在硝酸纖維膜(nc膜)上劃線為檢測線(t線),用0.1~0.3mg/ml(優選為0.2mg/ml)的羊抗兔二抗劃線作為控制線(c線),將金標墊裁剪為4~10mm(優選6mm)的寬度,待乾燥後組裝試紙條,使用免疫定量讀數儀度數。其中,所述mc-lr-ova為微囊藻毒素mc-lr與載體蛋白偶聯製得的人工抗原。所述新型小分子結構抗體標記的膠體金複合物由如下方法製備得到:將超純水加熱至沸騰,加入0.5~1.5%(優選1%)氯金酸,再次加熱至沸騰,加入2~5%(優選4%)檸檬酸三鈉,煮沸直至溶液顏色由藍紫色變為紫紅色且不再發生變化後2~5min;待膠體金溶液冷卻後,向膠體金溶液中加入0.1~0.3mol/ml(優選0.2mol/ml)碳酸鉀溶液震蕩,再加入的權利要求5所述抗體後將混合液靜置反應3~8min(優選5min),再加入15~25%(優選20%)bsa封閉,6000~10000rpm離心3~8min(優選8000rpm離心5min),溶液濃縮至原體積的85~90%。其中,優選地,所述超純水:1%氯金酸:4%檸檬酸三鈉的體積比為400:16:5~8。優選地,所述膠體金溶液:碳酸鉀溶液:抗體:bsa的體積比為1ml:1μl:2μl:20μl。另外,上述藍綠藻肝毒素廣譜免疫層析試紙條在檢測微囊藻毒素結構類似物和/或節球藻毒素方面的應用,也在本發明的保護範圍之內。優選地,所述微囊藻毒素結構類似物和/或節球藻毒素公有九種:mc-yr、mc-lr、mc-wr、nod、mc-la、mc-rr、mc-lw、mc-ly和mc-lf。本發明具有以下有益效果:(1)本發明設計了一種新型小分子結構lh-1((2e,4e,6s,7s)-7-甲氧基-4,6-二甲基-8-苯辛基-2,4-二烯酸)的合成方法,該方法簡單可行,可以用於與載體蛋白偶聯作為免疫原或包被原增加檢測方法的廣譜性;(2)本發明得到的lh-1抗體對mc-lr進行檢測的ic50為7.0ng/ml,lod為0.1ng/ml,ic20-ic80為0.6-87.8ng/ml,對mc-lw、mc-yr、mc-lf、mc-wr、nod、mc-la、mc-rr和mc-ly的交叉反應率分別為405.7%、164.6%、163.5%、136.6%、129.4%、92.1%、70.1%和37.8%,lod均低於1ng/ml故本法所得新型小分子結構lh-1多克隆抗體靈敏度良高,特異型好,可用於建立藍綠藻肝毒素免疫層析試紙條;(3)本發明製得的藍綠藻肝毒素廣譜性免疫層析試紙條靈敏度高,廣譜性好,可半定量檢測九種主要微囊藻毒素結構類似物和節球藻毒素,對其中的mc-yr、mc-lr、mc-wr、nod、mc-la檢測限可以達到1ng/ml,mc-rr、mc-lw、mc-ly和mc-lf檢測限也可達到2ng/ml,具有廣闊的發展前景。附圖說明圖1.廣譜性免疫層析試紙條製備流程圖。圖2.新型小分子結構lh-1合成過程示意圖。圖3.新型小分子結構lh-1人工抗原紫外掃描鑑定曲線。圖4.新型小分子結構lh-1抗體間接競爭elisa標準曲線。圖5.廣譜性免疫層析試紙條廣譜性檢測結果。圖6.廣譜性免疫層析試紙條基質效應的影響。具體實施方式以下結合說明書附圖和具體實施例來進一步說明本發明,但實施例並不對本發明做任何形式的限定。除非特別說明,本發明採用的試劑、方法和設備為本
技術領域:
常規試劑、方法和設備。除非特別說明,以下實施例所用試劑和材料均為市購。本發明研究由如下基金支持:國家自然科學基金:藻毒素廣譜特異性免疫識別分子基礎研究(u1301214);廣東省大學生科技創新培育專項資金:微囊藻毒素廣譜性快速檢測膠體金免疫層析試紙條的研製(pdjh2016b0089)。以下實施例包括新型小分子結構lh-1的合成、人工抗原的合成、新型小分子結構lh-1多克隆抗體的製備、新型小分子結構lh-1抗體的elisa檢測、藍綠藻肝毒素廣譜性免疫試紙條的製備,流程如圖1所示。實施例1新型小分子lh-1結構的合成1、新型小分子結構lh-1的合成新型小分子結構lh-1的合成路線圖如圖2所示,具體反應流程如下:(a)縮二脲醛醇縮合:室溫下,稱取26.2g右旋n-丙醯基硫偶氮烷硫酮(cas:1217320-19-8)溶於250ml無水二氯甲烷中攪拌,之後在丙酮中用乾冰冷卻至-78℃,逐滴向上述溶液中加入12ml四氯化鈦,所得混合物攪拌15min形成鈦絡合物,再向上述溶液中加入18.9ml蒸餾過的n,n-二異丙基乙胺,將混合物接著攪拌40min形成相應的烯醇化鈦,再加入20.0ml新蒸餾的n-甲基吡咯烷酮,10min後,加入13.4ml新蒸餾的苯乙醛,將上述溶液持續在-78℃再放置30min後溫熱至4℃再攪拌1h。通過tlc監測反應,展開劑為環己烷-乙酸乙酯(7:3,v/v),最後加入300ml50%nh4cl水溶液猝滅反應。將有機層用300ml1.0n的hcl和300ml10%na2co3和300ml鹽水洗滌兩次,再用na2so4乾燥。得到黃色固體狀的醛醇2。(b)反式醯胺化反應:將37.1g醛醇2溶於無水150ml二氯甲烷中,依次加入14.6gn,o-二甲基羥胺鹽酸鹽和27.2g咪唑,將此混合物在室溫下攪拌12h。通過tlc監測反應,展開劑為環己烷-乙酸乙酯(7:3,v/v)。然後用300ml去離子水將有機層洗滌3次,再用na2so4乾燥,過濾並真空濃縮。所得殘餘物通過矽膠柱層析純化,稱取600g矽膠填柱,以環己烷和乙酸乙酯(acoet,30-50%)作為流動相梯度洗脫,得到黃色油狀物中間體weinreb醯胺。(c)烷基化反應:將40.1ml碘甲烷溶於140ml無水四氫呋喃中,再將此溶液與1.38g固體na2co3混合攪拌10min後冷卻至4℃,通過脫脂棉過濾並加入到先前獲得的15.1gweinreb醯胺中,之後,在4℃下向上述溶液中加入2.49g的nah(60%分散在礦物油中),將反應混合物在室溫下攪拌4h。通過tlc監測反應,展開劑為環己烷-乙酸乙酯(6:4,v/v)。再加入1.24g的nah(60%油溶液),將反應混合物在室溫下再攪拌2h。通過tlc監測反應,展開劑為環己烷-乙酸乙酯(6:4,v/v)。冷卻至4℃後,過量的nah通過加入150ml飽和nh4cl去除。混合物用acoet萃取兩次,有機層再用250ml鹽水洗滌,之後用na2so4乾燥,過濾並減壓濃縮。用300g矽膠填柱後,用環己烷-acoet(6:4,v/v)作為流動相純化,得到黃色油狀weinreb醯胺3。(d)還原反應:將10.71gweinreb醯胺3溶於100ml無水二氯甲烷中,將所得溶液用在丙酮中的乾冰冷卻至-78℃。在80min內滴加100ml的1.0m二異丁基氫化鋁的無水甲苯溶液。通過tlc監測反應,展開劑為環己烷-乙酸乙酯(7:3,v/v),再依次加入40ml甲醇和40ml飽和nh4cl猝滅反應,之後將該溶液升溫至室溫攪拌30min。然後將混合物通過545墊,並用300ml苯乙基洗滌,合併有機相後用200ml2.0nhcl洗滌,再用mgso4乾燥,過濾並真空濃縮得到橙色油狀中間體醛。(e)wittig反應:將8.48g新製備的醛溶於275ml無水甲苯中,加入18.52g磷葉立德(1-乙氧基羰基亞乙基)三苯基正膦,將所得混合物回流反應12h。通過tlc監測反應,展開劑為環己烷-乙酸乙酯(8:2,v/v)。將反應混合物減壓濃縮,所得殘餘物通過矽膠柱純化,用0-10%acoet的環己烷溶液作為流動相,得到黃色油狀烯烴4。(f)還原反應:將7.59g烯烴4溶於55ml無水四氫呋喃中,並將所得溶液用丙酮中的乾冰冷卻至-78℃,在1h內向上述溶液滴加68ml1.0m二異丁基氫化鋁的無水甲苯溶液,在該溫度下攪拌90min,並通過tlc監測反應,展開劑為環己烷-乙酸乙酯(8:2,v/v),之後依次加入30ml甲醇和30mlnh4cl淬滅反應。使所得溶液升溫至室溫30min後將混合物用545墊過濾並用150ml二氯甲烷洗滌,再將有機層用200ml1.0nhcl洗滌,合併有機層並用mgso4乾燥,過濾並真空濃縮,再經過矽膠柱層析純化,以0-20%acoet的環己烷溶液作為流動相梯度洗脫,得到淡黃色油狀的烯丙醇。(g)氧化反應:將3.4g烯丙醇溶於100ml無水甲苯中,將所得溶液冷卻至0℃,再加入13.6gmno2,將所得懸浮液在55℃劇烈攪拌2h。通過tlc監測反應,展開劑為環己烷-乙酸乙酯(8:2,v/v),將所得溶液用545墊過濾,並用大量二氯甲烷洗滌。將所得濾液減壓濃縮,得到黃色油狀物醛5。(h)wittig反應:將3.39g新製備的醛5溶於80ml無水甲苯中,加入6.06g磷葉立德(1-乙氧基羰基亞甲基)三苯基正膦,將所得反應混合物攪拌並回流過夜。通過tlc監測反應,展開劑為環己烷-乙酸乙酯(8:2,v/v)。此後,將反應混合物用545墊過濾,用二氯甲烷洗滌並減壓濃縮。所得油狀殘餘物通過矽膠柱純化,以0-20%acoet的環己烷溶液作為流動相梯度洗脫,得到黃色油狀物酯6。(i)還原反應:將1.0g酯6溶於12ml無水四氫呋喃中,並將所得溶液用乾冰的丙酮冷卻至-78℃,在15min內向上述溶液中逐滴滴加12ml1.0m二異丁基氫化鋁的無水甲苯溶液,在-78℃下攪拌90min,並通過tlc監測反應,展開劑為環己烷-乙酸乙酯(8:2,v/v),之後在-78℃下,緩慢加入5ml甲醇和5mlnh4cl淬滅反應,然後將溶液溫熱至室溫攪拌30min。加入50ml1.0nhcl後,用acoet萃取有機相,並用mgso4乾燥,過濾並在真空下濃縮。所得殘餘物通過矽膠柱純化,以0-20%acoet的環己烷溶液作為流動相梯度洗脫,得到黃色油狀中間體醇。(j)氧化反應:將上一步所得醇全部溶於35ml無水甲苯中,冷卻至4℃。加入4.0gmno2,在50℃下攪拌1h。通過tlc監測反應,展開劑為環己烷-乙酸乙酯(7:3,v/v),並將懸浮液用545墊過濾,並用大量二氯甲烷洗滌,減壓濃縮濾液得到黃色油狀醛7。(k)氧化反應:將醛7溶於10mldmso中,再加入3.0g1,3,5-三甲氧基苯後攪拌5min,之後將2.0gnaclo2和3.0gnah2po4溶於水中,再將該水溶液逐滴加入到dmso溶液中並持續攪拌,通過tlc監測反應,展開劑為環己烷-乙酸乙酯(1:1,v/v),反應4h後使用na2s2o3猝滅反應並加入5ml1.0nhcl酸化反應,通過acoet萃取有機相併用水洗滌,再用mgso4乾燥,所得殘餘物通過矽膠柱純化,以40%acoet的正己烷溶液(0.1%hoac)作為流動相梯度洗脫,減壓濃縮濾液得到黃色油狀羧酸8,經tlc純化反應,展開劑為環己烷-乙酸乙酯(1:1,v/v)。2、鑑定:取新型小分子結構lh-1進行氫譜核磁、碳譜核磁和質譜鑑定。氫譜核磁結果如下:1h-nmr(400mhz,cdcl3)δh7.41(1h,d,j=10.4hz,h-2),7.28(2h,dd,j=8.8hz,j=2.8hz,h-3』,5』),7.17-7.22(3h,m,h-2』,4』,6』),5.88(1h,d,j=6.4hz,h-5),5.81(1h,d,j=10.4hz,h-3),3.27(3h,s,c7-och3),3.22-3.26(1h,m,h-7),2.80(1h,dd,j=9.2hz,j=3.2hz,h-8a),2.72(1h,dd,j=9.2hz,j=4.8hz,h-8b),2.63-2.68(1h,m,h-6),1.68(3h,s,c4-ch3),1.07(3h,d,j=4.4hz,c6-ch3)。碳譜核磁結果如下:13c-nmr(100mhz,cdcl3)δc172.9(c-1),152.0(c-3),145.7(c-1』),138.9(c-5),132.5(c-4),129.4(c-3』,5』),128.3(c-2』,6』),126.2(c-4』),115.3(c-2),86.4(c-7),58.7(c7-och3),38.1(c-8),37.1(c-6),15.6(c4-ch3),12.3(c6-ch3)質譜結果如下:esi-ms:m/z[m-h]-243.2(c17h21o3)。兩者結果皆表明合成結果正確且成功,成功合成得到新型小分子結構lh-1。實施例2人工抗原的合成1、人工抗原的合成(1)準確稱取2mglh-1,1mgnhs和1mgedc溶解於500μldmf中,室溫下避光過夜攪拌,未見沉澱出現;(2)8mg鑰孔血藍蛋白(klh)加入到500μl的碳酸緩衝液(0.1mol/l,ph9.6)中;(3)將lh-1溶液緩慢逐滴加入klh溶液中,室溫下避光攪拌3h;(4)用磷酸鹽緩衝液透析兩天,每天4次,透析結束後得lh-1人工抗原。(5)mc-lr與卵清蛋白(ova)的偶聯方法同上。碳酸鹽溶液(0.1m,ph9.6):稱量1.59gna2co3和2.93gnahco3,用去離子水定容1000ml。磷酸鹽緩衝液(0.01m,ph7.4):na2hpo4·12h2o2.90g,nacl8.50g,kcl0.20g,kh2po40.20g,加去離子水定容至1000ml。2、鑑定取人工抗原進行紫外掃描,結果如圖3所示。lh-1、klh和人工抗原分別進行紫外(200~400nm)掃描鑑定,並通過比較偶聯前後的各物質的最高吸光值,發現人工抗原的吸收曲線與載體蛋白明顯不同,klh僅在280nm處有特徵峰,lh-1在300nm處有特徵峰,而偶聯反應後,lh-1-klh在290nm處出現特徵峰,對比三者曲線可看出發生顯著位移,故說明反應產物是載體蛋白與lh-1的複合物,偶聯成功。實施例3新型小分子結構lh-1多克隆抗體的製備及純化1、新型小分子結構lh-1抗體製備用lh-1-klh免疫紐西蘭大白兔,將1mg/ml的人工抗原溶液與等量佐劑混合乳化,初次免疫與弗氏完全佐劑混合乳化後免疫,之後的加強免疫使用免疫原與弗氏不完全佐劑乳化後免疫,共免疫四次,每次間隔3周,每次免疫劑量為0.6ml/只,第四次免疫後第十天採血。2、新型小分子結構lh-1多克隆抗體純化(辛酸-硫酸銨法):(1)取兔血清,在4℃條件下,12000r/min離心15min,去除沉澱;(2)取1體積的兔血清與2體積的醋酸鹽緩衝液混合(ph4.8),室溫攪拌下,逐滴加入正辛酸,使用量為75μl正辛酸/ml兔血清;(3)室溫攪拌混合30min,4℃靜止2h使其充分沉澱;(4)4℃條件下,12000r/min離心15min,去除沉澱;(5)上清經砂芯漏鬥或125μm尼龍網過濾後,加入1/10體積的pbs緩衝液(0.1m,ph7.4),用2m的氫氧化鈉調ph至7.4,計算總溶液體積;(6)冰浴下於30min內加入0.277g/ml的硫酸銨,使其成為45%飽和溶液;(7)4℃靜止1h以上後,4℃條件下,12000r/min離心15min,棄上清;(8)沉澱用pbs緩衝液(0.1m,ph7.4)溶解並透析三天。實施例4新型小分子結構lh-1抗體的elisa檢測1、elisa檢測流程(1)將mc-lr-ova人工抗原用包被液稀釋至0.5μg/ml的濃度,包被96孔酶標板,每孔加入100μl,37℃溫育過夜;(2)棄去包被液,洗滌2次;(3)每孔加入120μl封閉液(5%脫脂奶粉),37℃封閉30min;(4)棄去封閉液,拍板,37℃烘乾備用;(5)用pbst1:8000倍稀釋新型小分子結構lh-1抗體,並將微囊藻毒素mc-lr稀釋至1000,100,10,1,0.1,0.01,0.001ng/ml;(6)每行加50μlmc-lr稀釋液(三組平行),再加50μl抗體稀釋液/孔,在37℃溫育40min,洗滌5次;(7)加入羊抗兔二抗-hrp(4000倍稀釋),37℃溫育30min,洗滌5次,拍板;(8)加入顯色液顯色10min;(9)加入50μl10%h2so4終止反應,並在450nm處讀取od值;(10)將mc-lr換成mc-rr、mc-yr、mc-lw、mc-lf、mc-la、mc-ly、mc-wr和nod八種藥物分別檢測,使用ic50的比值計算交叉反應率。2、檢測結果lh-1-klh免疫所得抗體間接競爭elisa標準曲線如圖4所示。對mc-lr的識別性能良好,其ic50為7.0ng/ml,lod為0.1ng/ml,ic20-ic80為0.6-87.8ng/ml,對mc-lw、mc-yr、mc-lf、mc-wr、nod、mc-la、mc-rr和mc-ly的交叉反應率分別為405.7%、164.6%、163.5%、136.6%、129.4%、92.1%、70.1%和37.8%,lod均低於1ng/ml故本法所得新型小分子結構lh-1多克隆抗體靈敏度良高,特異型好,可用於建立藍綠藻肝毒素免疫層析試紙條。實施例5藍綠藻肝毒素廣譜性免疫試紙條的製備1、膠體金的製備將400ml超純水加熱至沸騰,加入16ml的1%氯金酸,再次加熱至沸騰,加入5~8ml的4%檸檬酸三鈉,煮沸直至溶液顏色由藍紫色變為紫紅色且不再發生變化後2~5min,帶膠體金溶液冷卻後備用,所得金顆粒大小均一。2、新型小分子結構lh-1多抗膠體金的標記向1ml膠體金中分別加入1μl的0.2mol/ml碳酸鉀溶液震蕩,再加入2μl的多抗,將混合液靜置反應5min,再加入20μl的20%bsa封閉,8000rpm離心5min,濃縮至900μl,噴塗金標墊(60μl/cm2)。3、樣品墊液的確定用6mg/ml的mc-lr-ova在硝酸纖維膜(nc)上劃線為檢測線(t線),用0.2mg/ml的羊抗兔二抗劃線作為控制線(c線),將金標墊裁剪為6mm的寬度,待乾燥後組裝試紙條,用5‰caseinna+1‰、2‰、3‰、4‰吐溫+曲拉通10mmolpbs溶液做樣品液,點樣孔上分別滴加60μl的樣品液,每隔5min使用免疫定量讀數儀度數。結果如表1。表1時間1‰吐溫+曲拉通2‰吐溫+曲拉通3‰吐溫+曲拉通4‰吐溫+曲拉通5min6147.240.344.910min88.37072.857.115min117.789.1113.386.620min128106149.9115.8可得出使用5‰caseinna+1‰吐溫+曲拉通10mmolpbs溶液做樣品液時,顯色最快且顏色最深。4、mc-lr-ova包被量的確定用1、4、6、8mg/ml的mc-lr-ova在nc膜上劃線為檢測線(t線),用0.2mg/ml的羊抗兔二抗劃線作為控制線(c線),將金標墊裁剪為6mm的寬度,待乾燥後組裝試紙條,用5‰caseinna+1‰吐溫+曲拉通10mmolpbs溶液塗樣品墊,使用蒸餾水為陰性樣品,稀釋成1ng/ml的微囊藻毒素mc-lr作為陽性樣品,點樣時,每孔分別滴加60μl,每隔5min使用免疫定量讀數儀度數。結果如表2。表2可得出使用8mg/mlmc-lr-ova溶液包被於nc膜上時,顯色最快且顏色最深,且陰陽性數值差最明顯,且可看出在15min後反應趨於平衡,數值變化不明顯,故於15min時度數。5、試紙條靈敏度及廣譜性的檢測使用上述實施例中確定的條件噴塗nc膜、金標墊和樣品墊,組裝試紙條,將試紙條裁成3.5nm寬度,使用蒸餾水為陰性樣品,將mc-lr、mc-yr、mc-rr、mc-wr、mc-la、mc-lw、mc-ly、mc-lf和don分別稀釋成8、4、2、1、0.5、0.25、0.125、0.0625ng/ml作為陽性樣品進行檢測,點樣時,每孔分別滴加60μl,每隔5min使用免疫定量讀數儀度數,每組重複三次,結果如圖5所示。製得的藍綠藻肝毒素廣譜性免疫層析試紙條靈敏度高,廣譜性好,可半定量檢測九種主要藍綠藻肝毒素,對其中的mc-yr、mc-lr、mc-wr、nod、mc-la檢測限可以達到1ng/ml,mc-rr、mc-lw、mc-ly和mc-lf檢測限也可達到2ng/ml,具有廣闊的發展前景。6、試紙條基質效應的檢測使用上述實施例中確定的條件噴塗nc膜、金標墊和樣品墊,組裝試紙條,將試紙條裁成3.5nm寬度,分別用蒸餾水和湖水分別將mc-lr稀釋成8、4、2、1、0.5、0.25、0.125、0.0625,0ng/ml,點樣時,每孔分別滴加60μl,每隔5min使用免疫定量讀數儀度數,每組重複三次,結果如圖6所示,即本技術所述試紙條在檢測湖水時,檢測限仍可以達到1ng/ml,故該免疫層析試紙條可對經過濾後的湖水樣品進行檢測。當前第1頁12