一種合成C2‑乙醯氧基‑3‑吲哚酮類化合物及方法與流程
2023-04-29 18:26:17 3
本發明屬有機合成化學技術領域,具體涉及一種合成C2-乙醯氧基-3-吲哚酮類化合物及其方法。
背景技術:
3-吲哚酮同系物或衍生物是重要的雜環化合物,3-吲哚酮結構存在於許多藥物活性分子中,在材料化學、醫藥化學等領域應用廣泛。
常規的非吲哚直接氧化合成3-吲哚酮類的方法主要可以分為兩類:
一類為成環反應,一般以1-硝基-2-(苯基乙炔基)苯類似物的成環反應或N-(2-乙炔苯基)-4-甲基苯磺醯胺類似物的成環反應為主。此類反應的原料不易得,有些反應要用到貴金屬,或者各種複雜配體。如:2011年,Gagosz課題組以1-疊氮基-2-(丁-1-炔-1-基)苯為反應原料,選用複雜結構的貴金屬金催化劑,二氯乙烷做溶劑,成環生成有手性中心的3-吲哚酮類產物,產率可達到90%,雖然該反應得到的產率很高,需要昂貴的且含有複雜配體的金金屬的作為催化劑。(A.Wetzel and F.Gagosz,Angewandte Chemie,International Edition,2011,50,7354-7358.)。隨後,在2015年,浙江大學賈義霞教授課題組以1-硝基-2-(苯基乙炔基)苯為反應原料,貴金屬三氯化金作為催化劑,手性含膦酸作為配體,一定量分子篩下,甲苯作溶劑,生成了手性3-吲哚酮,雖然有較好的收率和光學活性,反應使用了貴重的金屬催化劑,及複雜的膦手性配體。這些都嚴重限制了其在藥物,工業上的發展。(R.-R.Liu,S.-C.Ye,C.-J.Lu,G.-L.Zhuang,J.-R.Gao and Y.-X.Jia,Angewandte Chemie,International Edition,2015,54,11205-11208.)。該類成環反應雖能較高產率地合成3-吲哚酮,卻不能用於對吲哚化合物的直接修飾,制約了此類反應的進一步應用與發展。
第二類為在原有簡單3-吲哚酮基本結構上構築新的複雜3-吲哚酮類產物。近年來,此類反應在藥物中間體,全合成前體,以及聚合物合成中得到極大的發展。2008年,耶魯大學Hamilton課題組以2,2-取代的二氫吲哚-3-酮為基本結構,反應生成了類似蛋白質結構的含3-吲哚酮的聚合物,證明了3-吲哚酮結構在聚合物中,有一定的運用前景。(P.N.W.a.A.D.Hamilton,Journal of the American Chemical Society,2009,131,4566–4567.)。在2015年,上海有機所Dai課題組以1-乙基-2-苄基二氫吲哚-3-酮為反應物,和烯丙基甲基碳酸酯,在貴金屬鈀做催化劑,複雜膦配體,二甲醚作溶劑,-40攝氏度下,得到了C2位具有手性中心的複雜3-吲哚酮結構。(T.-G.Chen,P.Fang,X.-L.Hou and L.-X.Dai,Synthesis,2015,47,134-140.)。雖然該類反應確實能較高產率的合成出帶有手性且複雜多樣的3-吲哚酮產物,但它們大都反應步奏繁瑣,需要複雜配體,及貴金屬。且所需底物本身含有3-吲哚酮基本結構,這些大大的限制了,此類方法的運用。因此,開發一種新型的、經濟適用且簡單高效地3-吲哚酮類化合物的合成方法具有十分重要的應用價值。
通過吲哚直接氧化方法對吲哚類化合物進行修飾直接合成3-吲哚酮類化合物現有已經公開的方法:
2014年,Xiao課題組報導了以吲哚衍生物3-苄基-2-苯基吲哚為底物,直接氧化生成2-苄基-2-苯基吲哚-3-酮的合成方法。反應條件中,使用了含有釕的光催化劑,氯仿作溶劑,整個反應體系在室溫,經LED燈的光照下,得以順利進行。產率在25%-76%。反應過程中,涉及到了,光催化及重排,這些對於產物的生成,起了極大的作用。(W.Ding,Q.-Q.Zhou,J.Xuan,T.-R.Li,L.-Q.Lu and W.-J.Xiao,Tetrahedron Letters,2014,55,4648-4652.)。2016年,Guchhait課題組同樣報導了吲哚直接氧化構造3-吲哚酮的新方法,並得到了較高的產率。(S.K.Guchhait,V.Chaudhary,V.A.Rana,G.Priyadarshani,S.Kandekar and M.Kashyap,Organic Letters,2016,18,1534-1537.)。上所述的合成方法需要使用昂貴的貴金屬作催化劑,或者複雜的配體,因此,發展一種更簡單高效的反應體系,來豐富與發展構造3-吲哚酮類化合物反應有著積極的意義。
技術實現要素:
本發明的目的在於提供一種成本低、簡單、綠色環保的合成C2-乙醯氧基-3-吲哚酮類化合物的方法。
為了實現上述目的,本發明的技術方案如下:一種合成C2-乙醯氧基-3-吲哚酮類化合物的的方法,式I所示的C2-乙醯氧基-3-吲哚酮類化合物
是通過式II所示的吲哚或取代吲哚。
與式III所示的2-氯嘧啶反應
生成的式IV所示的1-(嘧啶基-2-基)-1H-吲哚類化合物
在醋酸碘苯,乙酸/乙酸酐的參與下進行氧化成酮反應,其反應通式為:
其中,R基選自氫、烷基、烷氧基、苄氧基、滷代基或酯基,取代於吲哚環的C3、C4、C5、C6或C7位。
上述的一種合成C2-乙醯氧基-3-吲哚酮類化合物的方法,具體步驟如下:
將1-(嘧啶基-2-基)-1H-吲哚類化合物、醋酸碘苯溶於乙酸/乙酸酐混合溶劑中,混合均勻後在厚壁耐壓管中反應24-36小時,反應溫度設定為60℃,反應結束後,使用乙酸乙酯萃取出有機相,用飽和碳酸氫鈉水溶液除去溶劑,分離出有機相使用無水硫酸鈉除水,乾燥後使用減壓蒸餾除去溶劑得到粗產物,再粗產物經柱層析(15%EA/PE)分離後即得純產物取代C2-乙醯氧基-3-吲哚酮類化合物。
優選地,醋酸碘苯的摩爾量為1-(嘧啶基-2-基)-1H-吲哚類化合物摩爾量的3equiv.溶劑的量為乙酸/乙酸酐等於7比3,醋酸碘苯的摩爾體積比為0.25mol/L.。
優選地,1-(嘧啶基-2-基)-1H-吲哚類化合物通過如下步驟合成:
將吲哚或取代吲哚底物和NaH溶於N,N-二甲基甲醯胺(DMF)溶劑中,冰水浴條件下反應30~60分鐘,然後加入2-氯嘧啶,在100~130℃下反應16~24小時,反應結束後冷卻至室溫,使用乙酸乙酯萃取出有機相,用水除去溶劑,分離出有機相使用無水硫酸鈉除水,乾燥後使用減壓蒸餾除去溶劑得到粗產物,通過柱層析分離粗產物得到化合物1-(嘧啶基-2-基)-1H-吲哚類反應物。
優選地,1-(嘧啶基-2-基)-1H-吲哚類化合物合成步驟中所述的NaH的摩爾量為吲哚或取代吲哚摩爾量的1.2~1.8equiv.,2-氯嘧啶的摩爾量是吲哚或取代吲哚摩爾量的1.1~1.6equiv.。
優選地,取代吲哚中的取代基選自烷基、烷氧基、苄氧基、滷代基、滷素,取代於吲哚環的C3、C4、C5、C6、C7位。
附圖說明
圖1是實施例1製得的3-氧代-2-乙醯氧基-1-(嘧啶-2-基)吲哚的氫核磁譜圖(a)和碳核磁譜圖(b)。
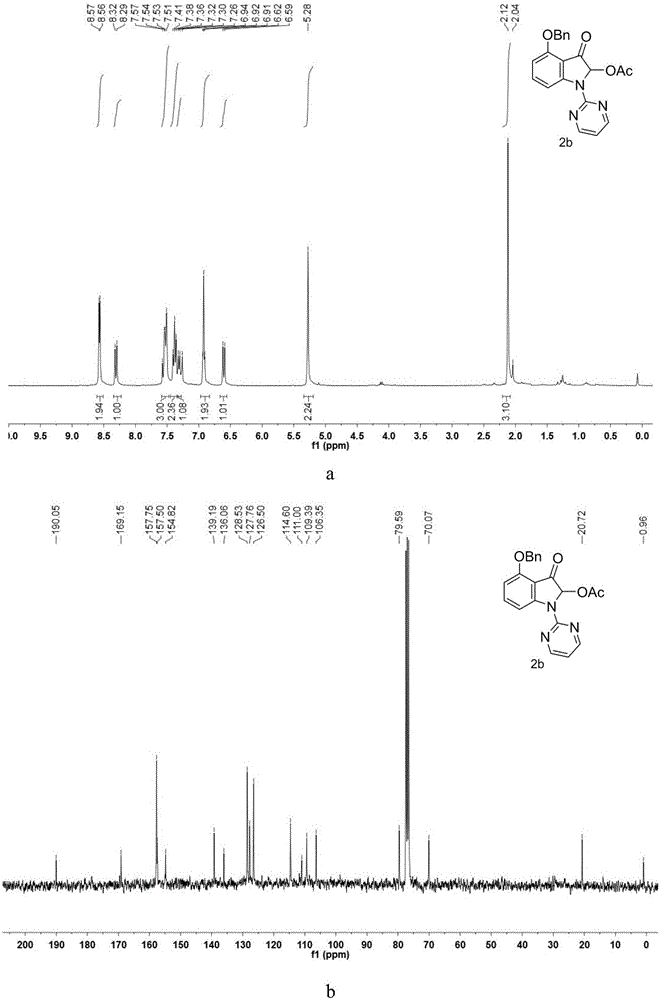
圖2是實施例2製得的4-(苄氧基)-2-乙醯氧基-3-氧代-1-(嘧啶-2-基)吲哚的氫核磁譜圖(a)和碳核磁譜圖(b)。
圖3是實施例3製得的5-甲基-2-乙醯氧基-3-氧代-1-(嘧啶-2-基)吲哚的氫核磁譜圖(a)和碳核磁譜圖(b)。
圖4是實施例4製得的4-甲基-2-乙醯氧基-3-氧代-1-(嘧啶-2-基)吲哚的氫核磁譜圖(a)和碳核磁譜圖(b)。
圖5是實施例5製得的6-氟-2-乙醯氧基-3-氧代-1-(嘧啶-2-基)吲哚的氫核磁譜圖(a)和碳核磁譜圖(b)。
圖6是實施例6製得的6-氯-2-乙醯氧基-3-氧代-1-(嘧啶-2-基)吲哚的氫核磁譜圖(a)和碳核磁譜圖(b)。
圖7是實施例7製得的5-溴-2-乙醯氧基-3-氧代-1-(嘧啶-2-基)吲哚的氫核磁譜圖(a)和碳核磁譜圖(b)。
圖8是實施例8製得的5-氟-2-乙醯氧基-3-氧代-1-(嘧啶-2-基)吲哚的氫核磁譜圖(a)和碳核磁譜圖(b)。
圖9是實施例9、10、11、12製得的5-氯-2-乙醯氧基-3-氧代-1-(嘧啶-2-基)吲哚的氫核磁譜圖(a)和碳核磁譜圖(b)。
與現有技術相比,本發明的顯著效果如下:
(1)採用廣泛易得、便宜的吲哚作為起始原料,運用吲哚直接氧化的反應方法,反應原子經濟性高,避免了常規的C-H的預活化。
(2)採用非金屬醋酸碘苯作為唯一氧化劑參與吲哚類底物的C2-乙醯氧基-3-吲哚酮反應,未使用昂貴貴金屬及複雜配體,顯著降低了成本。
(3)本發明合成的C2-乙醯氧基-3-吲哚酮類化合物可以對其進行產物運用,生成更為複雜結構,此結構單元具有廣泛的生物活性和廣闊的應用前景。
具體實施方式
以下通過列舉實例和附圖對本發明作進一步詳細說明。
實驗例1
3-氧代-2-乙醯氧基-1-(嘧啶-2-基)吲哚的合成
在反應容器中,加入吲哚(6mmol,702.9mg),NaH(質量分數60%,10.8mmol,432mg),DMF溶劑,低溫條件下反應30分鐘;然後加入2-氯嘧啶(9.6mmol,1099.488mg),混合物在130℃下反應24小時後,冷卻到室溫,萃取除去溶劑,過柱分離得到化合物1-(嘧啶基-2-基)-1H-吲哚反應物。用萬分之一電子天平準確稱量1-(嘧啶基-2-基)-1H-吲哚反應物(0.3mmol,58.50mg),轉移至反應容器厚壁耐壓管,向反應容器加入PhI(OAc)2(3eq,289.5mg)滴加2mL乙酸/乙酸酐=7/3混合溶劑,旋緊反應管塞使反應體系密封,加熱至60℃,油浴攪拌條件下反應36h。反應結束後,將反應液冷卻至室溫,使用一段短矽膠柱去除一些金屬雜質進行初步純化,萃取並除溶劑酸後,減壓旋蒸除去溶劑得到粗產物,粗產物進行柱層析分離(洗脫劑:乙酸乙酯/石油醚=15:75),得到純淨乾燥的產物,產率65%。
1H NMR(300MHz,CDCl3)δ8.74(d,J=8.4Hz,1H),8.59(d,J=4.8Hz,2H),7.76(d,J=7.6Hz,1H),7.68(s,1H),7.16(d,J=7.4Hz,1H),6.94(t,J=4.8Hz,1H),6.90(s,1H),2.13(s,3H).13C NMR(75MHz,CDCl3)δ193.17,169.18,157.86,157.63,153.73,137.61,124.36,122.76,121.98,117.24(s),114.56(s),79.54(s),20.65(s).IR(neat):2979,2848,1754,1723,1559,1461,1461,1428,1012,745.HRMS(ESI)calcd.for C14H11N3O3:269.0800found 269.0806。
實驗例2
4-(苄氧基)-2-乙醯氧基-3-氧代-1-(嘧啶-2-基)吲哚的合成
取100mL圓底燒瓶及大小適中的磁子,稱量4-(苄氧基)-1H-吲哚(5mmol,1115mg)溶解於N,N-二甲基甲醯胺(DMF)中,並置於冰水浴中緩慢加入NaH(質量分數60%,7.5mmol,300mg)進行拔氫反應,反應1個小時後轉移至130℃油浴,24h後通過TLC監測反應結束,用乙酸乙酯稀釋後轉移至250mL分液漏鬥,用水洗三遍,洗淨DMF後,收集有機相用無水硫酸鈉除水,減壓旋蒸除去有機溶劑後得到粗產物,通過柱層析(乙酸乙酯:石油醚=3:95)得到產物4-(苄氧基)-1H-(嘧啶-2-基)-1H-吲哚反應物。用萬分之一電子天平準確稱量4-(苄氧基)-1H-(嘧啶-2-基)-1H-吲哚反應物(0.3mmol,90.3mg),轉移至反應容器厚壁耐壓管中,向反應容器加入PhI(OAc)2(3eq,289.5mg)滴加乙酸/乙酸酐=7/3混合溶劑,旋緊反應管塞使反應體系密封,加熱至60℃,油浴攪拌條件下反應36h。反應結束後,將反應液冷卻至室溫,使用一段短矽膠柱去除一些金屬雜質進行初步純化,萃取並除溶劑酸後,減壓旋蒸除去溶劑得到粗產物,粗產物進行柱層析分離(洗脫劑:乙酸乙酯/石油醚=15:75),得到純淨乾燥的產物,產率71%。
1H NMR(300MHz,CDCl3)δ8.57(d,J=4.7Hz,2H),8.31(d,J=8.3Hz,1H),7.54(dd,J=11.4,7.9Hz,3H),7.38(t,J=7.4Hz,2H),7.31(d,J=7.2Hz,1H),6.95–6.89(m,2H),6.60(d,J=8.3Hz,1H),5.28(s,2H),2.12(s,3H).13C NMR(75MHz,CDCl3)δ190.05,169.15,157.74,154.50,139.19,136.06,128.52,127.76,126.49,114.60,110.99,109.39,106.35,79.59,70.06,20.71,0.95.IR(neat):2923,2851,1752,1715,1619,1584,1560,1450,1416,1146,191.HRMS(ESI)calcd.for C21H17N3O4:375.1219found 375.1226。
實驗例3
5-甲基-2-乙醯氧基-3-氧代-1-(嘧啶-2-基)吲哚的合成
取100mL圓底燒瓶及大小適中的磁子,稱量5-甲基-1H-吲哚(5mmol,755mg)溶解於N,N-二甲基甲醯胺(DMF)中,並置於冰水浴中緩慢加入NaH(質量分數60%,7.5mmol,300mg)進行拔氫反應,反應1個小時後轉移至130℃油浴,26h後通過TLC監測反應結束,用乙酸乙酯稀釋後轉移至250mL分液漏鬥,用水洗三遍,洗淨DMF後,收集有機相用無水硫酸鈉除水,減壓旋蒸除去有機溶劑後得到粗產物,通過柱層析(乙酸乙酯:石油醚=2:95)得到產物5-甲基-1H-(嘧啶-2-基)-1H-吲哚反應物。用萬分之一電子天平準確稱量5-甲基-1H-(嘧啶-2-基)-1H-吲哚反應物(0.3mmol,62.7mg),轉移至反應容器厚壁耐壓管中,向反應容器加入PhI(OAc)2(3eq,289.5mg)滴加乙酸/乙酸酐=7/3混合溶劑,旋緊反應管塞使反應體系密封,加熱至60℃,油浴攪拌條件下反應36h。反應結束後,將反應液冷卻至室溫,使用一段短矽膠柱去除一些金屬雜質進行初步純化,萃取並除溶劑酸後,減壓旋蒸除去溶劑得到粗產物,粗產物進行柱層析分離(洗脫劑:乙酸乙酯/石油醚=15:75),得到純淨乾燥的產物,產率70%。
1H NMR(300MHz,CDCl3)δ8.61(s,1H),8.57(d,J=4.8Hz,2H),7.56(s,1H),7.49(d,J=8.1Hz,1H),6.91(t,J=4.8Hz,1H),6.87(s,1H),2.38(s,3H),2.12(s,3H).13C NMR(75MHz,CDCl3)δ193.19,169.21,157.76,151.88,138.62,132.58,124.10,117.05,114.27,79.83,20.64,20.50.IR(neat):2924,2850,1756,1715,1621,1581,1560,1417,1191,1146,788.HRMS(ESI)calcd.for C15H13N3O3:283.0957found 283.0962。
實驗例4
4-甲基-2-乙醯氧基-3-氧代-1-(嘧啶-2-基)吲哚的合成
取100mL圓底燒瓶及大小適中的磁子,稱量4-甲基吲哚(5mmol,656mg)溶解於N,N-二甲基甲醯胺(DMF)中,並置於冰水浴中緩慢加入NaH(質量分數60%,7.5mmol,300mg)進行拔氫反應,反應1個小時後轉移至130℃油浴,24h後通過TLC監測反應結束,用乙酸乙酯稀釋後轉移至250mL分液漏鬥,用水洗三遍,洗淨DMF後,收集有機相用無水硫酸鈉除水,減壓旋蒸除去有機溶劑後得到粗產物,通過柱層析(乙酸乙酯:石油醚=3:95)得到產物4-甲基-1H-(嘧啶-2-基)-1H-吲哚反應物。用萬分之一電子天平準確稱量4-甲基-1-(嘧啶基-2-基)-1H-吲哚反應物((0.3mmol,62.7mg),轉移至反應容器厚壁耐壓管中,向反應容器加入PhI(OAc)2(3eq,289.5mg)滴加乙酸/乙酸酐=7/3混合溶劑,旋緊反應管塞使反應體系密封,加熱至60℃,油浴攪拌條件下反應36h。反應結束後,將反應液冷卻至室溫,使用一段短矽膠柱去除一些金屬雜質進行初步純化,萃取並除溶劑酸後,減壓旋蒸除去溶劑得到粗產物,粗產物進行柱層析分離(洗脫劑:乙酸乙酯/石油醚=15:75),得到純淨乾燥的產物,產率65%。
1H NMR(300MHz,CDCl3)δ8.65–8.46(m,3H),7.63–7.35(m,1H),6.92(dd,J=6.2,3.4Hz,1H),6.89(d,J=6.6Hz,2H),2.62(d,J=8.5Hz,3H),2.11(d,J=9.9Hz,3H).13C NMR(75MHz,CDCl3)δ193.76(s),169.22(s),157.73(s),154.17(s),139.95(s),136.84(s),124.63(s),119.78(s),114.42(s),79.39(s),20.68(s),18.29(s).IR(neat):2931,2849,1745,1718,1597,1574,1558,1410,1207,779.HRMS(ESI)calcd.for C15H13N3O3:283.0957found 283.0949。
實驗例5
6-氟-2-乙醯氧基-3-氧代-1-(嘧啶-2-基)吲哚的合成
取100mL圓底燒瓶及大小適中的磁子,稱量6-氟吲哚(5mmol,675mg)溶解於N,N-二甲基甲醯胺(DMF)中,並置於冰水浴中緩慢加入NaH(質量分數60%,7.5mmol,300mg)進行拔氫反應,反應1個小時後轉移至130℃油浴,24h後通過TLC監測反應結束,用乙酸乙酯稀釋後轉移至250mL分液漏鬥,用水洗三遍,洗淨DMF後,收集有機相用無水硫酸鈉除水,減壓旋蒸除去有機溶劑後得到粗產物,通過柱層析(乙酸乙酯:石油醚=3:95)得到產物6-氟-1H-(嘧啶-2-基)-1H-吲哚反應物。用萬分之一電子天平準確稱量6-氟-1-(嘧啶基-2-基)-1H-吲哚反應物((0.3mmol,63.9mg),轉移至反應容器厚壁耐壓管中,向反應容器加入PhI(OAc)2(3eq,289.5mg)滴加乙酸/乙酸酐=7/3混合溶劑,旋緊反應管塞使反應體系密封,加熱至60℃,油浴攪拌條件下反應36h。反應結束後,將反應液冷卻至室溫,使用一段短矽膠柱去除一些金屬雜質進行初步純化,萃取並除溶劑酸後,減壓旋蒸除去溶劑得到粗產物,粗產物進行柱層析分離(洗脫劑:乙酸乙酯/石油醚=15:75),得到純淨乾燥的產物,產率60%。
1H NMR(300MHz,CDCl3)δ8.60(d,J=4.8Hz,2H),8.49(dd,J=11.0,2.2Hz,1H),7.76(dd,J=8.4,5.9Hz,1H),6.98(t,J=4.8Hz,1H),6.89(s,1H),6.88–6.79(m,1H),2.12(s,3H).13C NMR(75MHz,CDCl3)δ191.25,169.03,168.81(d,J=255.7Hz),157.87,157.46,155.41(d,J=14.9Hz),126.49(d,J=11.8Hz),118.43,114.99,110.72(d,J=24.5Hz),105.04(d,J=29.7Hz),79.86,20.58.IR(neat):2919,2850,1748,1717,1618,1576,1555,1456,1435,793.HRMS(ESI)calcd.for C14H10FN3O3:287.0706found 287.0709.
實驗例6
6-氯-2-乙醯氧基-3-氧代-1-(嘧啶-2-基)吲哚的合成
取100mL圓底燒瓶及大小適中的磁子,稱量6-氯吲哚(5mmol,755mg)溶解於N,N-二甲基甲醯胺(DMF)中,並置於冰水浴中緩慢加入NaH(質量分數60%,7.5mmol,300mg)進行拔氫反應,反應1個小時後轉移至130℃油浴,24h後通過TLC監測反應結束,用乙酸乙酯稀釋後轉移至250mL分液漏鬥,用水洗三遍,洗淨DMF後,收集有機相用無水硫酸鈉除水,減壓旋蒸除去有機溶劑後得到粗產物,通過柱層析(乙酸乙酯:石油醚=3:95)得到產物6-氯-1H-(嘧啶-2-基)-1H-吲哚反應物。用萬分之一電子天平準確稱量6-氯-1-(嘧啶基-2-基)-1H-吲哚反應物((0.3mmol,68.7mg),轉移至反應容器厚壁耐壓管中,向反應容器加入PhI(OAc)2(3eq,289.5mg)滴加乙酸/乙酸酐=7/3混合溶劑,旋緊反應管塞使反應體系密封,加熱至60℃,油浴攪拌條件下反應36h。反應結束後,將反應液冷卻至室溫,使用一段短矽膠柱去除一些金屬雜質進行初步純化,萃取並除溶劑酸後,減壓旋蒸除去溶劑得到粗產物,粗產物進行柱層析分離(洗脫劑:乙酸乙酯/石油醚=15:75),得到純淨乾燥的產物,產率59%。
1H NMR(300MHz,CDCl3)δ8.80(d,J=1.4Hz,1H),8.61(d,J=4.8Hz,2H),7.67(d,J=8.2Hz,1H),7.12(dd,J=8.2,1.4Hz,1H),6.97(d,J=4.8Hz,1H),6.87(s,1H),2.12(s,3H).13C NMR(75MHz,CDCl3)δ191.76,169.02,157.88,157.46,154.05,144.00,125.14,123.31,120.46,117.51,114.99,79.75,20.57.IR(neat):2997,2840,1750,1732,1603,1574,1554,1417,1065,786.HRMS(ESI)calcd.for C14H10ClN3O3:303.0411found 303.0405.
實驗例7
5-溴-2-乙醯氧基-3-氧代-1-(嘧啶-2-基)吲哚的合成
取100mL圓底燒瓶及大小適中的磁子,稱量5-溴吲哚(5mmol,975mg)溶解於N,N-二甲基甲醯胺(DMF)中,並置於冰水浴中緩慢加入NaH(質量分數60%,7.5mmol,300mg)進行拔氫反應,反應1個小時後轉移至130℃油浴,24h後通過TLC監測反應結束,用乙酸乙酯稀釋後轉移至250mL分液漏鬥,用水洗三遍,洗淨DMF後,收集有機相用無水硫酸鈉除水,減壓旋蒸除去有機溶劑後得到粗產物,通過柱層析(乙酸乙酯:石油醚=3:95)得到產物5-溴-1H-(嘧啶-2-基)-1H-吲哚反應物。用萬分之一電子天平準確稱量5-溴-1-(嘧啶基-2-基)-1H-吲哚反應物((0.3mmol,81.9mg),轉移至反應容器厚壁耐壓管中,向反應容器加入PhI(OAc)2(3eq,289.5mg)滴加乙酸/乙酸酐=7/3混合溶劑,旋緊反應管塞使反應體系密封,加熱至60℃,油浴攪拌條件下反應36h。反應結束後,將反應液冷卻至室溫,使用一段短矽膠柱去除一些金屬雜質進行初步純化,萃取並除溶劑酸後,減壓旋蒸除去溶劑得到粗產物,粗產物進行柱層析分離(洗脫劑:乙酸乙酯/石油醚=15:75),得到純淨乾燥的產物,產率57%。
1H NMR(300MHz,CDCl3)δ8.63(d,J=8.9Hz,1H),8.58(d,J=4.8Hz,2H),7.82(d,J=1.9Hz,1H),7.71(dd,J=8.9,2.1Hz,1H),6.96(t,J=4.8Hz,1H),6.81(s,1H),2.11(s,3H).13C NMR(75MHz,CDCl3)δ191.85,169.02,157.87,157.54,152.40,139.85,126.79,123.65,118.95,115.55,114.84,79.66,20.55.IR(neat):2921,2852,1734,1574,1463,1232,1044,733.HRMS(ESI)calcd.for C14H10BrN3O3:346.9906found 346.9909。
實驗例8
5-氟-2-乙醯氧基-3-氧代-1-(嘧啶-2-基)吲哚的合成
取100mL圓底燒瓶及大小適中的磁子,稱量5-氟吲哚(5mmol,675mg)溶解於N,N-二甲基甲醯胺(DMF)中,並置於冰水浴中緩慢加入NaH(質量分數60%,7.5mmol,300mg)進行拔氫反應,反應1個小時後轉移至130℃油浴,24h後通過TLC監測反應結束,用乙酸乙酯稀釋後轉移至250mL分液漏鬥,用水洗三遍,洗淨DMF後,收集有機相用無水硫酸鈉除水,減壓旋蒸除去有機溶劑後得到粗產物,通過柱層析(乙酸乙酯:石油醚=3:95)得到產物5-氟-1H-(嘧啶-2-基)-1H-吲哚反應物。用萬分之一電子天平準確稱量5-氟-1-(嘧啶基-2-基)-1H-吲哚反應物((0.3mmol,63.9mg),轉移至反應容器厚壁耐壓管中,向反應容器加入PhI(OAc)2(3eq,289.5mg)滴加乙酸/乙酸酐=7/3混合溶劑,旋緊反應管塞使反應體系密封,加熱至60℃,油浴攪拌條件下反應36h。反應結束後,將反應液冷卻至室溫,使用一段短矽膠柱去除一些金屬雜質進行初步純化,萃取並除溶劑酸後,減壓旋蒸除去溶劑得到粗產物,粗產物進行柱層析分離(洗脫劑:乙酸乙酯/石油醚=15:75),得到純淨乾燥的產物,產率56%。
1H NMR(300MHz,CDCl3)δ8.80–8.70(m,1H),8.58(d,J=4.8Hz,2H),7.41(d,J=7.7Hz,2H),6.95(s,1H),6.85(s,1H),2.13(s,3H).13C NMR(75MHz,CDCl3)δ192.47,169.09,157.84,157.64,157.19(d,J=245.7Hz),150.06(s),124.56(d,J=23.6Hz),123.06(d,J=7.5Hz),118.81(d,J=6.5Hz),114.62,109.86(d,J=23.3Hz),80.06,20.56.IR(neat):2979,1754,1727,1621,1579,1556,1418,1210,1133,882.HRMS(ESI)calcd.for C14H10FN3O3:287.0706found 287.0703.
實驗例9
5-氯-2-乙醯氧基-3-氧代-1-(嘧啶-2-基)吲哚的合成
取100mL圓底燒瓶及大小適中的磁子,稱量5-氯吲哚(5mmol,755mg)溶解於N,N-二甲基甲醯胺(DMF)中,並置於冰水浴中緩慢加入NaH(質量分數60%,7.5mmol,300mg)進行拔氫反應,反應1個小時後轉移至130℃油浴,24h後通過TLC監測反應結束,用乙酸乙酯稀釋後轉移至250mL分液漏鬥,用水洗三遍,洗淨DMF後,收集有機相用無水硫酸鈉除水,減壓旋蒸除去有機溶劑後得到粗產物,通過柱層析(乙酸乙酯:石油醚=3:95)得到產物5-氯-1H-(嘧啶-2-基)-1H-吲哚反應物。用萬分之一電子天平準確稱量5-氯-1-(嘧啶基-2-基)-1H-吲哚反應物((0.3mmol,68.7mg),轉移至反應容器厚壁耐壓管中,向反應容器加入PhI(OAc)2(3eq,289.5mg)滴加乙酸/乙酸酐=7/3混合溶劑,旋緊反應管塞使反應體系密封,加熱至60℃,油浴攪拌條件下反應36h。反應結束後,將反應液冷卻至室溫,使用一段短矽膠柱去除一些金屬雜質進行初步純化,萃取並除溶劑酸後,減壓旋蒸除去溶劑得到粗產物,粗產物進行柱層析分離(洗脫劑:乙酸乙酯/石油醚=15:75),得到純淨乾燥的產物,產率55%。
1H NMR(300MHz,CDCl3)δ8.69(d,J=8.9Hz,1H),8.58(d,J=4.8Hz,2H),7.68(s,1H),7.58(d,J=8.9Hz,1H),6.96(t,J=4.8Hz,1H),6.83(s,1H),2.11(s,3H).13C NMR(75MHz,CDCl3)δ192.0,169.05,157.87,157.4,152.01,137.09,128.35,123.71,123.23,118.59,114.82,79.81,20.55.IR(neat):2922,2846,1752,1733,1669,1771,1561,1456,834,700.HRMS(ESI)calcd.for C14H10ClN3O3:303.0411found 303.0401。
實驗例10
5-氯-2-乙醯氧基-3-氧代-1-(嘧啶-2-基)吲哚的合成
取100mL圓底燒瓶及大小適中的磁子,稱量5-氯吲哚(5mmol,755mg)溶解於N,N-二甲基甲醯胺(DMF)中,並置於冰水浴中緩慢加入NaH(質量分數60%,7.5mmol,300mg)進行拔氫反應,反應1個小時後轉移至130℃油浴,24h後通過TLC監測反應結束,用乙酸乙酯稀釋後轉移至250mL分液漏鬥,用水洗三遍,洗淨DMF後,收集有機相用無水硫酸鈉除水,減壓旋蒸除去有機溶劑後得到粗產物,通過柱層析(乙酸乙酯:石油醚=3:95)得到產物5-氯-1H-(嘧啶-2-基)-1H-吲哚反應物。用萬分之一電子天平準確稱量5-氯-1-(嘧啶基-2-基)-1H-吲哚反應物((0.3mmol,68.7mg),轉移至反應容器厚壁耐壓管中,向反應容器加入PhI(OAc)2(3eq,289.5mg)滴加乙酸/乙酸酐=7/3混合溶劑,旋緊反應管塞使反應體系密封,加熱至40℃,油浴攪拌條件下反應36h。反應結束後,將反應液冷卻至室溫,使用一段短矽膠柱去除一些金屬雜質進行初步純化,萃取並除溶劑酸後,減壓旋蒸除去溶劑得到粗產物,粗產物進行柱層析分離(洗脫劑:乙酸乙酯/石油醚=15:75),得到純淨乾燥的產物,產率為50%。
1H NMR(300MHz,CDCl3)δ8.69(d,J=8.9Hz,1H),8.58(d,J=4.8Hz,2H),7.68(s,1H),7.58(d,J=8.9Hz,1H),6.96(t,J=4.8Hz,1H),6.83(s,1H),2.11(s,3H).13C NMR(75MHz,CDCl3)δ192.0,169.05,157.87,157.4,152.01,137.09,128.35,123.71,123.23,118.59,114.82,79.81,20.55.IR(neat):2922,2846,1752,1733,1669,1771,1561,1456,834,700.HRMS(ESI)calcd.for C14H10ClN3O3:303.0411found 303.0401。
實驗例11
5-氯-2-乙醯氧基-3-氧代-1-(嘧啶-2-基)吲哚的合成
取100mL圓底燒瓶及大小適中的磁子,稱量5-氯吲哚(5mmol,755mg)溶解於N,N-二甲基甲醯胺(DMF)中,並置於冰水浴中緩慢加入NaH(質量分數60%,7.5mmol,300mg)進行拔氫反應,反應1個小時後轉移至130℃油浴,24h後通過TLC監測反應結束,用乙酸乙酯稀釋後轉移至250mL分液漏鬥,用水洗三遍,洗淨DMF後,收集有機相用無水硫酸鈉除水,減壓旋蒸除去有機溶劑後得到粗產物,通過柱層析(乙酸乙酯:石油醚=3:95)得到產物5-氯-1H-(嘧啶-2-基)-1H-吲哚反應物。用萬分之一電子天平準確稱量5-氯-1-(嘧啶基-2-基)-1H-吲哚反應物((0.3mmol,68.7mg),轉移至反應容器厚壁耐壓管中,向反應容器加入PhI(OAc)2(3eq,289.5mg)滴加乙酸/乙酸酐=7/3混合溶劑,旋緊反應管塞使反應體系密封,加熱至80℃,油浴攪拌條件下反應36h。反應結束後,將反應液冷卻至室溫,使用一段短矽膠柱去除一些金屬雜質進行初步純化,萃取並除溶劑酸後,減壓旋蒸除去溶劑得到粗產物,粗產物進行柱層析分離(洗脫劑:乙酸乙酯/石油醚=15:75),得到純淨乾燥的產物,產率為53%。
1H NMR(300MHz,CDCl3)δ8.69(d,J=8.9Hz,1H),8.58(d,J=4.8Hz,2H),7.68(s,1H),7.58(d,J=8.9Hz,1H),6.96(t,J=4.8Hz,1H),6.83(s,1H),2.11(s,3H).13C NMR(75MHz,CDCl3)δ192.0,169.05,157.87,157.4,152.01,137.09,128.35,123.71,123.23,118.59,114.82,79.81,20.55.IR(neat):2922,2846,1752,1733,1669,1771,1561,1456,834,700.HRMS(ESI)calcd.for C14H10ClN3O3:303.0411found 303.0401。
實驗例12
5-氯-2-乙醯氧基-3-氧代-1-(嘧啶-2-基)吲哚的合成
取100mL圓底燒瓶及大小適中的磁子,稱量5-氯吲哚(5mmol,755mg)溶解於N,N-二甲基甲醯胺(DMF)中,並置於冰水浴中緩慢加入NaH(質量分數60%,7.5mmol,300mg)進行拔氫反應,反應1個小時後轉移至130℃油浴,24h後通過TLC監測反應結束,用乙酸乙酯稀釋後轉移至250mL分液漏鬥,用水洗三遍,洗淨DMF後,收集有機相用無水硫酸鈉除水,減壓旋蒸除去有機溶劑後得到粗產物,通過柱層析(乙酸乙酯:石油醚=3:95)得到產物5-氯-1H-(嘧啶-2-基)-1H-吲哚反應物。用萬分之一電子天平準確稱量5-氯-1-(嘧啶基-2-基)-1H-吲哚反應物((0.3mmol,68.7mg),轉移至反應容器厚壁耐壓管中,向反應容器加入PhI(OAc)2(3eq,289.5mg)滴加乙酸/乙酸酐=7/3混合溶劑,旋緊反應管塞使反應體系密封,加熱至60℃,油浴攪拌條件下反應24h。反應結束後,將反應液冷卻至室溫,使用一段短矽膠柱去除一些金屬雜質進行初步純化,萃取並除溶劑酸後,減壓旋蒸除去溶劑得到粗產物,粗產物進行柱層析分離(洗脫劑:乙酸乙酯/石油醚=15:75),得到純淨乾燥的產物,產率為52%。
1H NMR(300MHz,CDCl3)δ8.69(d,J=8.9Hz,1H),8.58(d,J=4.8Hz,2H),7.68(s,1H),7.58(d,J=8.9Hz,1H),6.96(t,J=4.8Hz,1H),6.83(s,1H),2.11(s,3H).13C NMR(75MHz,CDCl3)δ192.0,169.05,157.87,157.4,152.01,137.09,128.35,123.71,123.23,118.59,114.82,79.81,20.55.IR(neat):2922,2846,1752,1733,1669,1771,1561,1456,834,700.HRMS(ESI)calcd.for C14H10ClN3O3:303.0411found 303.0401。