氮氧化物作為新型組蛋白去乙醯化酶抑制劑的抗腫瘤應用的製作方法
2023-05-13 21:12:36 2
本發明涉及醫藥技術領域,尤其是涉及氮氧化物作為新型組蛋白去乙醯化酶抑制劑的製備、用途及其衍生物,包含此化合物的藥物組合物,涉及所述化合物的製備方法及所述化合物的抗腫瘤應用,涉及所述化合物在疾病,例如抗腫瘤細胞的應用。
背景技術:
hdac抑制劑已經在臨床上用於治療腫瘤疾病,並表現出良好的臨床應用前景。其hdac抑制劑對腫瘤細胞主要作用是抑制腫瘤細胞增殖,誘導細胞周期阻滯,誘導腫瘤細胞凋亡和抑制腫瘤新血管的生成等。研究表明hdac抑制劑既可以單獨用於治療腫瘤疾病,也可以與很多其他作用機制的抗腫瘤藥物聯合使用,都表現出明顯的抗腫瘤作用。hdac抑制劑作為一類新型的抗腫瘤藥物具有非常重要的研發價值。
目前,hdac抑制劑主要分為五類:異羥肟酸(hydroxamicacid)類,如saha、環氧酮結構的環四肽類如trapoxina、苯甲醯胺(benzamides)類,如ms-275、不含環氧酮結構的環肽類、短鏈和芳香脂肪酸類,如丁酸鈉、苯丁酸鈉、雜環化合物類,如depudecin。其中已經上市的hdac抑制劑包vorinostat(saha,zolina)、romidepsin(istodax,fk228,fr901228,depsipeptide、belinostat(beleodaq,pxd-101)、panobinostat(lbh-589,farydak)、西達本胺(chidamide)。根據已經上市的hdac抑制劑的結構特點,其主要包含鋅離子鰲合部分(zbg)、連接部分(linker)和表面部分(surfacerecognition,cap)三個部分。zbg部分與活性口袋底部的鋅離子進行配位,通常為異羥肟酸、苯甲酞胺、梭酸或親電酮等。linker主要佔據疏水管狀通道,以保證其他兩部分的正確取向,通常為鏈狀烷烴、肉桂酞基、芳香基或雜環芳香基等。cap與結合口袋入口處的胺基酸殘基緊密接觸,產生相互作用。以saha為例,其結構如圖3所示.
隨著分子生物學研究的不斷深入,hdac抑制劑的抗腫瘤作用機制逐漸被揭示,但對hdacs亞型是否具有選擇性,提高hdac抑制劑的體內穩定性等問題。因此,設計合成氮氧化物作為新型組蛋白去乙醯化酶抑制劑,以已經上市的結構特點為基礎,對zbg、linker部分進行修飾,以期發現對hdac亞型具有選擇性的hdac抑制劑以及對別的分子靶標的相互作用等。選擇抗腫瘤活性的化合物進一步的進行hdac亞型的選擇以及對他分子靶標的相互作用。因此,根據羥肟酸類化合物的結構特點進行修飾,主要是探索zbg,linker的變化導致的活性變化。氮氧化合物作為一類新的hdac抑制劑,以期發現對hdac亞型具有良好選擇性的化合物,為hdac抑制劑的後續靶向藥物開發提供重要理論依據,從而開發出藥效更好,特異性更強、選擇性更好的hdac抑制劑。
技術實現要素:
本發明的目的是通過氮氧化物作為新型組蛋白去乙醯化酶抑制劑,來尋找對hdac亞型具有良好的選擇性的抑制劑。本發明實現該目的的技術方案是提供氮氧化物作為新型組蛋白去乙醯化酶抑制劑及藥物組合的製備方法和應用,解決hdac抑制劑對hdac亞型選擇性等問題。
為解決上述技術問題,本發明採用下述技術方案:
本發明的氮氧化物作為新型組蛋白去乙醯化酶抑制劑,該化合物的結構通式如下:
其中:n可為1-10個碳;可為不飽和鍵。
x、z:可同時為c、n;如x=z=n或x=z=c,也可x=c、z=n或x=n、z=c;
r1、r2:c-r11、s、n、o當r1=c時r2可為c、s、n、o其中之一,當r1=c時r2可為c、s、n、o其中之一;
r3、r4:分別為苯環或雜環,其取代基可以為滷素、氨基、羥基、硝基、氰基、1-4個碳原子的烷基、1-4個碳原子的烷氧基、1-4個碳原子的烷氨基、1-4個碳原子的氨烷基、2-4個碳原子的醯基、2-4個碳原子的醯胺基、1-4個碳原子的硫代烷基、1-4個碳原子的全氟烷基、1-4個碳原子的全氟烷或雜環取代基;或分別為氫、羥基;或可同時為二甲胺也可當其中一個為氫或羥基時,另一個為二甲胺(接上二甲胺後再經過氧化得氮氧化合物);
r5:為羥基等;
r6、r7、r8、r9、r10:可以同時有1-4個取代基;當其中之一為滷素、氨基、羥基、硝基、氰基、磺醯基、烷氧基、醯基、2-4個碳原子的醯基、2-4個碳原子的醯胺基、1-4個碳原子的硫代烷基、1-4個碳原子的全氟烷基、1-4個碳原子的氟烷氧基、1-4個碳原子的羧基、1-4個碳原子的烷氧基羧基時;其餘分別為滷素、氨基、羥基、硝基、氰基、磺醯基、烷氧基、醯基、2-4個碳原子的醯基、2-4個碳原子的醯胺基、1-4個碳原子的硫代烷基、1-4個碳原子的全氟烷基、1-4個碳原子的氟烷氧基、1-4個碳原子的羧基、1-4個碳原子的烷氧基羧基;其中r6、r7、r8、r9、r10也可以為苯環或雜環;
其中,所述的雜環,是指含一個或多個雜原子(包括氮、氧、硫)的飽和或不飽和雜環,如四氫吡啶、二氫吡諾、二氫吡唑、哌啶、咪唑、吡啶等;
所述的滷素,為氟、氯、溴、碘;
所述的1-4個碳原子的烷基,包括甲基、乙基、正丙基、異丙基、正丁基、異丁基等;
所述的1-4個碳原子的烷氧基,包括甲氧基、乙氧基、正丙氧基、正丁氧基、異丁氧基等;
所述的1-4個碳原子的氨基烷基,包括氨基乙基、1-氨基丙基、1-氨基丙基等;
所述的1-4個碳原子的烷基胺基,包括n-甲胺基、n-乙胺基、n-異丙胺基等;
所述的2-4個碳原子的醯基,包括乙醯基、丙醯基、異丁醯基等;
所述的2-4個碳原子的醯胺基,包括乙醯胺基、丙醯胺基、丁醯胺基、異丁醯胺基等;
所述的1-4個碳原子的硫代烷基,包括甲硫基、乙硫基、丙硫基等;
所述的1-4個碳原子的全氟烷基,包括三氟甲基、五氟乙基等;以氮氧化物作為新型組蛋白去乙醯化酶抑制劑的製備方法包括步驟如下:
步驟1:為醯胺反應。在鹼性試劑作用下,使用苯胺與不同鏈長度的溴化物反應,製備獲得2a‐c,其中n=4、6、8;所述的鹼性試劑為diea,催化劑為dcc,溶劑為四氫呋喃,反應溫度為室溫,反應時間約5小時;
步驟2:2a‐c分別與l-脯氨醇、二苯基脯氨醇反應下之別獲得3a‐f,其中r=h、ph;採用k2co3使反應處於鹼性條件,加入適量nai可讓反應產率提高,反應體系採用dmf作為溶劑。反應溫度為50-70℃,約反應6個小時;
步驟3:此步為氧化反應,化合物3a‐f在氧化劑的作用下,製備獲得目標化合物4a‐f,即式(i)化合物,反應溫度為冰浴反應,溶劑為四氫呋喃,氧化劑為mcpba,時間約為3個小時。
其中,實驗本採用化合物2b、2c的合成方法來合成2a,但總是不成功,因此本發明改進將4-溴丁酸變為4-溴丁醯氯再與苯胺反應得到2a,反應式如下:2a的化合物為的製備方法如下:
本發明製得的氮氧化物作為新型組蛋白去乙醯化酶抑制劑對人肺癌細胞(a549),人乳腺癌細胞(mcf7)等多種腫瘤細胞生長抑制均較好,可應用於製備預防或治療腫瘤的藥物。
附圖說明
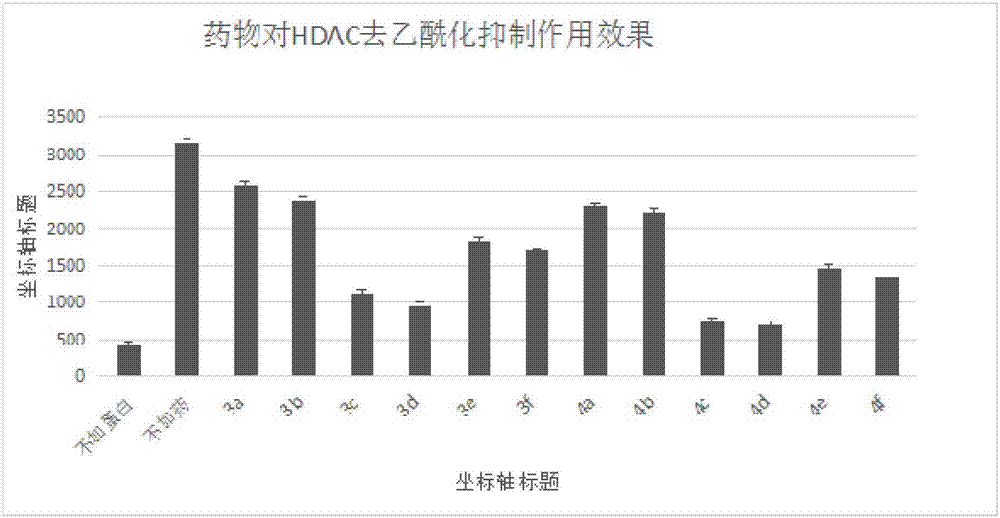
圖1是hdac抑制劑的篩選示意圖;
圖2是胰酶抑制劑的篩選示意圖;
圖3是saha結構示意圖。
具體實施方式
下面結合附圖和實施例對本發明的技術方案作進一步的詳細說明。
氮氧化物作為新型組蛋白去乙醯化酶抑制劑,具有以下結構式(i)、式(ⅱ)化合物:
其中:r選自h或ph;n=4、6、8。
式(i)化合物的製備方法包括如下步驟:
步驟1:為醯胺反應。在鹼性試劑作用下,使用苯胺與不同鏈長度的溴化物反應,製備獲得2a‐c,其中n=4、6、8;所述的鹼性試劑為diea,催化劑為dcc,溶劑為四氫呋喃,反應溫度為室溫,反應時間約5小時;
步驟2:2a‐c分別與l-脯氨醇、二苯基脯氨醇反應下之別獲得3a‐f,其中r=h、ph;採用k2co3使反應處於鹼性條件,加入適量nai可讓反應產率提高,反應體系採用dmf作為溶劑。反應溫度為50-70℃,約反應6個小時;
式(ⅱ)化合物的製備方法包括如下步驟:
步驟1:為醯胺反應。在鹼性試劑作用下,使用苯胺與不同鏈長度的溴化物反應,製備獲得2a‐c,其中n=4、6、8;所述的鹼性試劑為diea,催化劑為dcc,溶劑為四氫呋喃,反應溫度為室溫,反應時間約5小時;
步驟2:2a‐c分別與l-脯氨醇、二苯基脯氨醇反應下之別獲得3a‐f,其中r=h、ph;採用k2co3使反應處於鹼性條件,加入適量nai可讓反應產率提高,反應體系採用dmf作為溶劑。反應溫度為50-70℃,約反應6個小時;
步驟3:此步為氧化反應,化合物3a‐f在氧化劑的作用下,製備獲得目標化合物4a‐f,即式(i)化合物,反應溫度為冰浴反應,溶劑為四氫呋喃,氧化劑為mcpba,時間約為3個小時。
其中,化合物2a反應式和製備方法如下:
實驗本採用化合物2b、2c的合成方法來合成2a,但屢次不成功,因此改進將4-溴丁酸變為4-溴丁醯氯再與苯胺反應得到2a,冰浴反應,時間約為4小時,溶劑為四氫呋喃。
本發明化合物抗腫瘤活性的實驗方法與結果如下:
(1)組蛋白去乙醯化酶為靶標的氮氧化合物的抗腫瘤試劑對組蛋白去乙醯化酶活性的測定,採用螢光方法檢測:
培養a549細胞,從中提取核蛋白,獲取含hdac的活性生物樣本,進行活性測試,步驟如下:
實驗方法:
hdac抑制劑的篩選
在ep管中加入反應體系需要的溶液(兩控制組也要加入相應的dmso),最後加核提取物37℃孵育1h,(第一個樣品加入後間隔30s,再加入第二個樣品中),反應完成後,30s取一個插冰上,再加入2×胰酶繼續孵育1h,反應完成後,30s取一個插冰上,再取120ul加入96孔板中,360nm,460nm處測螢光。藥物濃度為100um/ml,部分數據見圖1。
從實驗結果中可以看出3c、3d、4c、4d在藥物濃度為100um的時候對hdac的抑制效果較好,可能存在潛在的hdac抑制劑。由於本實驗涉及兩步反應,不確定是抑制了hdac還是胰酶,所以下一步針對是否是胰酶的方面的原因,進行了胰酶抑制篩選的實驗。
胰酶抑制劑的篩選:
在ep管中加入反應體系需要的溶液(兩控制組也要加入相應的dmso),胰酶50ul(不加蛋白組加50ul水),37℃孵育1h(第一個樣品加入後間隔30s,再加入第二個樣品中),反應完成後,30s取一個插冰上,再取120ul加入96孔板中,在360nm,460nm處使用酶標儀檢測螢光。藥物濃度為100um/ml。部分數據見圖2。
根據如圖結果所示,雖然胰酶對該批藥物有輕微抑制作用,但是經過圖1圖2對比可知該批藥物對hdac有抑制作用。
(2)氮氧化物作為新型組蛋白去乙醯化酶抑制劑在抗腫瘤細胞的活性測試
採用公認的可用於大規模抗腫瘤藥物篩選、細胞毒性試驗測定的四甲基氮唑藍比色法(mtt)評價目標化合物對5種人癌細胞株的抗增殖活性。陽性對照藥為臨床上廣泛使用的抗腫瘤藥物阿黴素(adr)及vorinostat(saha)。
細胞株:人肺癌細胞a549,人乳腺癌細胞mcf7,人肝癌細胞hepg2,人結腸癌細胞sw480,腦膠質瘤細胞u87,宮頸癌細胞hela。
細胞增殖抑制率=(陰性對照組od值—藥物組od值)*100%/陰性對照組od值。
表1為本發明化合物對腫瘤細胞增殖的抑制率%(100μmol/l)部分實驗測試結果如表一:
*:saha的濃度為10μmol/l;其餘化合物濃度為100μmol/l。
由實驗數據可知所有化合物與陽性saha對照,所有的化合物的抑制劑率都相對偏低,其中3a、3b、4a、4b、3e、3f、4e、4f對a549、mcf7、hepg2、u87、hela腫瘤細胞抑制效率較其他化合物相對較好;3a、3b、3c、3d、3e、3f對a549、mcf7、hepg2、u87、hela腫瘤細胞抑制效率與陽性對照藥物saha的抑制率相對偏低,但與saha的抑制率相近。3a、3b、3c、3d、3e、3f分別與4a、4b、4c、4d、4e、4f相比抑制劑率相對偏低,說明氧化後的化合物相對未氧化的化合物抑制劑率較好;4a、4b、4c、4d、4e、4f的抑制率相比,說明linker的長短影響化合物的抑制劑率。故該類化合物具有較大的發展潛力。進一步考察linker部分的長短及結構修飾,有望能得到活性較好的化合物。
綜上所述,本發明的優點在於:本發明提供了一種氮氧化合物作為新型hdac抑制劑的抗腫瘤應用及其製備方法,根據活性測試實驗數據顯示:該類化合物對hdac有潛在抑制劑效果;因此,可進一步在zbg部分提供更多的氮氧、linker進行多個長度的考察等在現有的基礎上進行結構修飾,以期發現對hdac亞型具有良好選擇性的抑制劑及其他靶點的作用。本發明合成原料相對便宜、工藝簡便、純度較高、成本較低等優點,有望成為對hdac亞型選擇性強、高效低毒的新型hdac抑制劑類抗腫瘤藥物。
當然,以上只是本發明的具體應用範例,本發明還有其他的實施方式,凡採用等同替換或等效變換形成的技術方案,均落在本發明所要求的保護範圍之內。