一種可光交聯型二茂鐵基表面活性劑膠束的調控方法與流程
2023-05-07 04:35:32 1
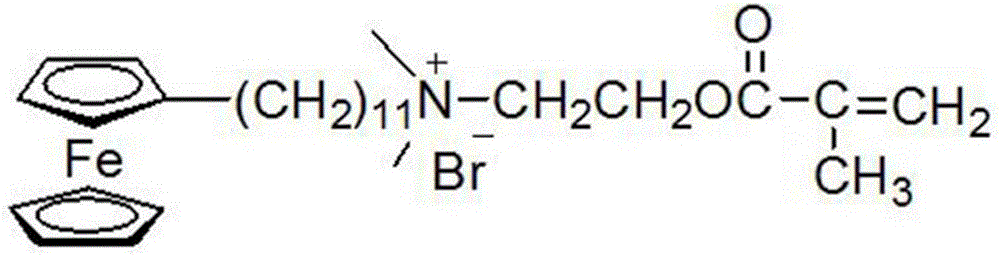
一種可光交聯型二茂鐵基表面活性劑膠束的調控方法,特別涉及一種能夠氧化還原反應的可光交聯型二茂鐵基表面活性劑膠束的調控方法。
背景技術:
二茂鐵表面活性劑是一類具有可逆氧化還原性質的兩親分子,其最大的特點是氧化態和還原態的CMC相差很大,如圖1所示,使得其在溶液中聚集形成的膠束或囊泡能隨氧化還原發生可逆的解離與重新聚集。利用這一特性,Tsuchiya等設計了一種電化學可控的自組裝體系,他們發現在水楊酸鈉溶液中陽離子二茂鐵表面活性劑可形成蠕蟲狀膠束,使體系的粘度顯著增大,氧化後,膠束結構被破壞,溶液粘度顯著降低,通過氧化還原,膠束結構和粘度能可逆地變化,實現了電化學控制的可逆聚集與解聚集。Li等則將二茂鐵季銨鹽與環糊精進行包合,然後與陰離子表面活性劑自組裝設計了一種氧化還原的可控自組裝體系。這些充分顯示了二茂鐵表面活性劑在可控自組裝領域的潛能,如何構建新的基於二茂鐵表面活性劑的可控自組裝體系是一個令人感興趣的課題,有待於進一步發展。
技術實現要素:
為了解決現有技術的問題,本申請合成了一種可聚合性二茂鐵表面活性劑,1.一種可光交聯型二茂鐵基表面活性劑膠束的調控方法,所述活性劑分子式為:11-二茂鐵基正十一烷基甲基丙烯酸乙酯基二甲基溴化銨,所述活性劑的水溶液可以形成球狀膠束,氧化後,球狀膠束解離,這一過程可逆,並且可以進一步通過光原位交聯來調控。所述活性劑的水楊酸鈉水溶液可以形成蠕蟲膠束,氧化後,蠕蟲膠束解離,這一過程伴隨粘度的巨大差異
可優選的是,所述活性劑通過甲基丙烯酸二甲胺基乙酯與11-溴代正十一烷基二茂鐵的親核取代反應合成。
可優選的是,所述11-二茂鐵基正十一烷基甲基丙烯酸乙酯基二甲基溴化銨具有氧化還原活性。
可優選的是,I+的臨界膠束濃度(CMC)為0.3mM,當I2+低於100mM時平衡表面張力仍隨著濃度的增大而下降。
可優選的是,所述球狀膠束形成及解離具體為:將1mM I+水溶液中加入單體量1wt%DMPA引發劑,加入單體量2wt%交聯劑DAP,直接紫外光聚合,獲得直徑為12nm(用的DLS數據,DLS數據與原子力數據的差異性來源於後者是乾燥後樣品測試結果,變大是因為膠束出現坍塌,直徑變大,不過平行比較仍有價值的球狀膠束,將上述體系加入1.1倍摩爾量的硫酸鐵,氧化後,DLS(動態光散射)觀察,通過原子力觀察,觀察到溶脹後的球狀膠束(DLS結果:溶脹球狀膠束直徑為17nm),此時,膠束未被解離,只是溶脹。
可優選的是,所述球狀膠束形成及解離具體為:將1mM I+水溶液中加入單體量1wt%DMPA引發劑,不加交聯劑DAP,直接紫外光聚合,獲得直徑為8nm的膠束;將上述體系加入1.1倍摩爾量的硫酸鐵,氧化後,DLS(動態光散射)觀察,通過原子力觀察,未觀察到膠束或囊泡的存在,說明膠束在氧化後發生解離。
可優選的是,I+的水楊酸鈉水溶液在一定濃度範圍可以形成蠕蟲膠束,體系粘度明顯上升;氧化後,蠕蟲膠束解離形成小的膠束,粘度隨之下降。
可優選的是,所述I+的水楊酸鈉水溶液為100mM的I+水溶液與20mM的水楊酸鈉水溶液混合。
可優選的是,所述活性劑的分子結構式為:
可優選的是,所述活性劑具有良好的電化學可逆性,可以用於化學傳感、電催化、修飾電極以及其他活性領域。
本申請首次合成了可聚合型二茂鐵表面活性劑I+。該表面活性劑具有良好的電化學可逆性,可以用於化學傳感,電催化,修飾電極等潛在應用。通過平衡表面張力與濃度的依賴性,發現I+的稀溶液氧化前後的表面活性存在明顯差異,利用這點,成功通過氧化、交聯等方式成功實現了膠束的調控。I+的水楊酸鈉水溶液可以形成蠕蟲膠束,氧化後,蠕蟲膠束解離,這一過程伴隨粘度的巨大差異。
附圖說明
圖1為陽離子二茂鐵表面活性劑自組裝膠束(左)與解組裝(右)示意圖。
圖2為可聚合型二茂鐵表面活性劑(I+)分子結構式示意圖
圖3為(11-二茂鐵基正十一烷基)(甲基丙烯酸乙酯基)二甲基溴化銨(I+)的合成路線示意圖。
圖4為室溫下化合物I+氧化前後的紫外光譜(氧化劑:硫酸鐵)。
圖5為I+的化學結構式以及其電子轉移反應式。
圖6為室溫下1mM I+在0.01M NaBr水溶液的CV圖(掃描速率為0.01V/s)。
圖7為平衡表面張力γ與濃度的依賴性:I+(●,還原態),I2+(○,氧化態)。
圖8為不同條件下I+水溶液(C=1.0mM)聚集體的尺寸分布。
圖9為不同條件下I+水溶液(C=1.0mM)原子力照片.a:不加DAP,氧化前;b:不加DAP,氧化後;c:2wt%DAP,氧化前;d:2wt%DAP,氧化後。
圖10為不加DAP,氧化前,I+水溶液(C=1.0mM)紫外光聚合後的原子力分析界面。
圖11為2wt%DAP,氧化前,I+水溶液(C=1.0mM)紫外光聚合後的原子力分析界面。
圖12為2wt%DAP,I+水溶液(C=1.0mM)紫外光聚合後,再氧化後的原子力分析界面。
圖13為氧化前後I+的水楊酸鈉水溶液(100mM的I+水溶液與20mM的水楊酸鈉水溶液混合)氧化前後粘度隨剪切速率的變化。
圖14為氧化還原調控體系粘度的變化。
具體實施例
如圖1所示為陽離子二茂鐵表面活性劑自組裝膠束(左)與解組裝(右)示意圖,其中,藍色和紫色球分別代表還原及氧化態二茂鐵基。
本申請首次合成了一種可聚合性二茂鐵表面活性劑(I+),其結構式如圖2所示所示,並進行了一系列表徵。
11-二茂鐵基正十一烷基甲基丙烯酸乙酯基二甲基溴化銨(I+)按照圖3所示的路線合成。
11-二茂鐵基正十一烷基甲基丙烯酸乙酯基二甲基溴化銨(I+)的合成是通過甲基丙烯酸二甲胺基乙酯與11-溴代正十一烷基二茂鐵的親核取代反應完成。具體來說:在裝有磁力攪拌子的100mL三口燒瓶中,加入甲基丙烯酸乙酯基二甲胺基乙酯1.6g(10.1mmol),11-溴代正十一烷基二茂鐵3.2g(7.6mmol),對苯二酚16mg,溶解於40mL丙酮中,在氬氣保護下,回流反應,TLC追蹤反應進程,72小時後反應結束。熱過濾,濾液濃縮,進而進行柱層析,流動相:二氯甲烷/甲醇=10:1,獲得3.1g金黃色固體,產率為70%。Rf=0.8(展開劑二氯甲烷/甲醇=5:1)。
I+的化學氧化還原,可聚合型二茂鐵表面活性劑I+可以通過化學(電化學)方法實現還原態(I+)和氧化態(I2+)的可逆轉換。通常來說,對化合物的氧化還原方法主要有化學氧化還原和電化學氧化還原。理論上上述兩種方法能取得相同的氧化還原效果。考慮到電化學方法完全氧化還原所需時間較長,我們採用了化學方法,具體來說,所有的二茂鐵系統,一般加入1.1倍摩爾量的氧化劑硫酸鐵氧化,還原時是加入1.1倍摩爾量的還原劑抗壞血酸。圖4是室溫下化合物I+氧化前後的紫外光譜(氧化劑:硫酸鐵),從圖可以看出,二茂鐵表面活性劑水體系在還原態時為黃色液體(最大吸收波長:440nm),氧化後為藍色液體(最大吸收波長:628nm)。
I+的紫外光聚合,在I+的稀水溶液(1mg/mL)中,加入紫外光引發劑2,2-二甲氧基-2-苯基乙醯苯(DMPA,單體的1wt%),和光交聯劑鄰苯二甲酸二烯丙酯(DAP,單體的0or2wt%),混合體系氬氣鼓泡除氧30分鐘,用橡皮塞密封。紫外光光照進行聚合(光強3mW/cm2),聚合過程通過1H NMR光譜監控:I+的重水體系中,雙鍵的質子峰完全消失時,聚合過程完成。一般聚合時間為5分鐘。
測試與表徵,傅立葉變換紅外光譜(FT-IR)採用Bruker Vertor 33傅立葉變換紅外光譜儀測試,構造單元及複合物均採用室溫下KBr壓片法。
核磁共振氫譜(1H NMR)、碳譜(13C NMR)採用德國Bruker公司Avance400核磁共振儀測量。
複合物氮含量N(wt%)用德國ELEMENTAR公司Vario EL元素分析儀測量。
X線衍射(XRD)採用飛利浦公司X』pert PRO型X射線衍射儀室溫下測試,Cu-Kα射線(λ=0.154nm),Ni片濾波,WAXD的掃描範圍為2θ=1-30°。測試時,掃描步長為Δ2θ=0.01°,掃描速率為2s/step。
紫外-可見光譜採用Hitachi公司UV-3010型紫外/可見光分光光度計測量。
示差掃描量熱分析(DSC)在Netzsch DSC 204上測量,氮氣保護,變溫速率為10℃/min,溫度變化順序為室溫140℃→-60℃→140℃→-60℃→140℃,第一次升溫的目的為消除樣品熱歷史。
熱重分析(TG)在Netzsch TG 209上測量,氮氣保護,升溫速率為10°/min,升溫範圍為室溫→800℃。
偏光顯微鏡(POM)使用的是Zeiss Axiophot型偏光顯微鏡,帶有附件Linkam熱臺。
表面張力測試採用K11model型表面張力儀,測試溫度:20.0±0.1℃,測試結果取3次平均值。
粒徑測試採用英國Malvern公司的Nano-ZS90Zeta電位和粒度分析儀於25℃測定,雷射波長為633nm,測試角度為90°。取2mL經0.22μm濾膜過濾的聚合物溶液加入到樣品池中,樣品在室溫下靜置10min後分別測定光散射強度和相關函數。
循環伏安法採用CHI-660電化學工作站測量,三電極體系:Φ3mm的玻碳電極作為工作電極,213型鉑片電極作為對電極,飽和甘汞電極作為參比電極,加入0.01M NaBr水溶液作為支持電解質。測試前,通高純氮除氧30分鐘,掃描範圍:-0.4~+1.0V,掃描速度0.01~4V/s,實驗在室溫下進行。
電化學性質,I+的化學結構式以及其電子轉移反應式如圖5所示。
二茂鐵有趣的氧化還原活性可以用於化學傳感,電催化,修飾電極等潛在應用。我們通過循環伏安曲線研究了I+在0.01M NaBr水溶液電化學行為。圖6為1mM I+在室溫下0.01M NaBr水溶液中的CV圖,掃描速率為0.01V/s,掃描電勢範圍為-0.05~+0.3V。從圖中可以看出,I+具有一對氧化還原峰,氧化峰電勢Epa為0.165V;還原峰電勢Epc為0.093V。那麼I+的氧化還原峰電勢差ΔE(=Epa-Epc)分別均為72mV。CV圖中氧化還原峰電勢差ΔE能夠反映電化學可逆性。I+的氧化還原峰電勢差ΔE小,可逆程度高。
膠束行為,圖7為I+與I2+的平衡表面張力對濃度的依賴性曲線,從圖中可以計算出I+的CMC(臨界膠束濃度)為0.3mM,與文獻報導的類似二茂鐵季銨鹽型表面活性劑FTMA的CMC(0.1mM)相近;當I2+低於100mM時平衡表面張力仍隨著濃度的增大而下降,說明其在我們所觀察濃度範圍內並未形成膠束。這是因為當二茂鐵基團被氧化成二茂鐵陽離子後,二茂鐵陽離子之間的靜電排斥作用增強,有利於二茂鐵表面活性劑分子的分散,同時二茂鐵基團由疏水基變成親水基,表面活性劑性質也隨之發生變化。在類似分子FTMA上也有同樣的現象。
在動態光散射中,利用快速數字時間相關儀記錄散射光強隨時間的漲落,即時間相關函數,可以得到散射光的特徵衰減時間,進而求得平均擴散係數D和與之相對應的流體力學半徑Rh。對於溶液中的納米粒子而言,它的流體力學半徑可以直接反映出聚集體在溶液中的大小。因此,動態光散射是研究高聚物溶液及膠體和粗分散體系的一種有效方法。
圖8為不同的聚合或是否氧化等條件下,1mM I+水溶液的粒徑分布圖。首先我們將1mM I+水溶液中加入單體量1wt%DMPA引發劑,不加交聯劑DAP,直接紫外光聚合,獲得直徑為8nm的膠束,其原子力照片如圖1-9a,通過原子力分析軟體計算其平均直徑為17nm(圖10),與DLS數據的誤差可能來自製樣的差別,原子力樣品需要真空乾燥,為固態形貌,但這並不影響我們研究其變化規律。我們將上述體系加入1.1倍摩爾量的硫酸鐵,氧化後,DLS觀察,流體力學直徑為26nm,通過原子力觀察,未觀察到膠束或囊泡的存在(如圖9b),說明膠束在氧化後可能發生解離。
我們將1mM I+水溶液中加入單體量1wt%DMPA引發劑,加入單體量2wt%交聯劑DAP,紫外光聚合,獲得直徑為12nm的膠束(圖8),其原子力照片如圖1-9c,通過原子力分析軟體計算其平均直徑為34nm(圖11),這說明,加入DAP後,聚合所得膠束粒徑有所增大。我們將上述體系加入1.1倍摩爾量的硫酸鐵,氧化後,DLS觀察,流體力學直徑為17nm。氧化後二茂鐵變成親水,使得膠束溶脹。通過原子力觀察,觀察到溶脹後的膠束,其原子力照片如圖1-9d,通過原子力分析軟體計算其平均直徑為48nm(圖12),比氧化前AFM計算出的34nm增大了14nm。
綜上,未加DAP,膠束未被交聯,氧化後,膠束解離。加入DAP後,膠束被交聯,氧化後,膠束未被解離,只是溶脹。上述結論為我們調控膠束提供了很好的方法。蠕蟲膠束,研究發現在一定的濃度範圍內,I+的水楊酸鈉水溶液氧化前後粘度的剪切速率依賴性有明顯差別。舉例來說,將100mM的I+水溶液與20mM的水楊酸鈉水溶液混合後,發現粘度明顯增加;但當加入氧化劑硫酸鐵後,粘度又馬上變小。
圖13為氧化前後I+的水楊酸鈉水溶液(100mM的I+水溶液與20mM的水楊酸鈉水溶液混合)氧化前後粘度隨剪切速率的變化,結合國外Abe教授的研究成果,他發現FTMA二茂鐵表面活性劑也存在類似現象,其結合TEM觀察發現,I+的水楊酸鈉水溶液在一定濃度範圍可以形成蠕蟲膠束,體系粘度明顯上升;氧化後,蠕蟲膠束解離形成小的膠束,粘度隨之下降。我們合成的I+的粘度變化與FTMA類似,也認為其形成了蠕蟲膠束,氧化後蠕蟲膠束解離。模型如圖14所示。
首次合成了可聚合型二茂鐵表面活性劑I+。該表面活性劑具有良好的電化學可逆性,可以用於化學傳感,電催化,修飾電極等潛在應用。通過平衡表面張力與濃度的依賴性,發現I+的稀溶液氧化前後的表面活性存在明顯差異,利用這點,我們成功通過氧化、交聯等方式成功實現了膠束的調控。I+的水楊酸鈉水溶液可以形成蠕蟲膠束,氧化後,蠕蟲膠束解離,這一過程伴隨粘度的巨大差異。