一種相選擇性凝膠劑及其製備方法和應用與流程
2023-04-30 14:59:01 2
本發明屬於功能材料技術領域,涉及一種相選擇性凝膠劑及其製備方法和應用。
背景技術:
油類洩漏對於環境和水域生態系統有著災難性的影響,而且對社會帶來重大的經濟損失。治理油類洩漏的傳統手段有吸附劑、分散劑、生物降解和固化劑等,但在實際應用中的效果不大。近期,越來越多的人對發現新型治理油類洩漏方法投以關注。其中最有前途的一種方法是用相選擇性有機凝膠因子來使水中的油形成凝膠。這種方法不僅有效且對環境友好。但是,通常需要大量的載體溶劑來均勻分散凝膠分子。這些溶劑,有的是溶於水的非凝膠溶劑,會擴散到水中;有的則是不溶於水的凝膠溶劑,會和油一起形成凝膠。在這兩種情況下,或者會給生態系統帶來影響,或者不利於油類的回收。因此,將凝膠因子以粉末的形式直接應用會更加實際且理想。
CN105686971A公開了一種聚胺基酸凝膠劑及其製備方法和應用,所述聚胺基酸凝膠劑使用以質量百分數計的1-8%的-聚穀氨酸、1-4%的聚賴氨酸、3-18%的-環糊精、70-95%的水以及餘量的功能成分製備而成,其主要用於面膜或者乳液等護膚品中作為添加劑,而並沒有膠凝油類的作用。
CN104803850A公開了一種酯類糖基相選擇性親油凝膠因子及其製備方法以及在油品膠凝中的應用,該發明以甘露糖醇和中長基鏈醯氯為原料採用O-酯化反應製備得到酯類糖基相選擇性親油凝膠因子,其可以進入油相,起到膠凝的作用。
CN105111108A公開了一種脂肪類叔胺雙脲小分子凝膠劑及其觸變性分子凝膠,該凝膠劑分子具有以下結構:
其中R代表H或C1-C5烷基;R1-R4各自獨立的代表H或C1-C2烷基。雖然該凝膠劑具有形成剪切觸變凝膠的能力,但是只有將其在有機溶劑中加熱溶解,而後冷卻才可以起到形成穩定的剪切觸變凝膠,其可用於製備有機或無機材料的無缺陷晶體的良性介質,但卻不能用於室溫下的油水分離。
因此,在本領域期望得到一種能夠直接以粉末的形式與水中的油類或有機溶劑形成凝膠的脲基有機凝膠因子。
技術實現要素:
針對現有技術的不足,本發明的目的在於提供一種相選擇性凝膠劑及其製備方法和應用。本發明的凝膠劑可以粉末的形式與水中的油類或有機溶劑形成凝膠,在室溫下完成油水分離。
為達此目的,本發明採用以下技術方案:
一方面,本發明提供一種相選擇性凝膠劑,所述相選擇性凝膠劑具有式I所示結構:
式I中,R為C8-C18(例如C8、C9、C10、C11、C12、C13、C14、C15、C16、C17或C18)的直鏈烷基,優選C12直鏈烷基(即-(CH2)11CH3)。
本發明的式I所示凝膠劑在油水混合物中可以選擇性進入油相,在油相中實現自組裝,形成物理交聯的超分子聚合物,可以使得油類(如汽油、煤油、柴油、機油)以及非極性的有機溶劑(例如正己烷、環己烷、苯、甲苯或二甲苯)形成凝膠,是一種具有相選擇性的凝膠劑。可以粉末的形式與水中的油類或有機溶劑形成凝膠,在室溫下完成油水分離。
通常在兩個脲基或醯胺基之間連上平面的芳香基團,使π-π相互作用和氫鍵共同作用形成超分子聚合物,因此認為如果在兩個脲基之間引入彎曲的橋連雙萘可能會影響π-π鍵和氫鍵這兩種作用的共同作用,但是事實情況卻並不是這樣,本發明的相選擇性凝膠劑中兩個脲基通過橋連的雙萘骨架連接在一起後,兩個脲基可以促進橋連雙萘骨架之間的非共價相互作用,使得式I所示化合物具有膠凝劑性質。然而,如果在兩個醯胺基之間引入彎曲的橋連雙萘卻不會產生這樣的效果。
在兩個脲基通過橋連的雙萘骨架連接在一起形成式I所示凝膠劑,橋連的雙萘骨架在固體狀態下,以反向平行的方式形成「從背後擁抱」的類似於「超分子聚合物」的結構,相鄰分子間存在的C-H…π相互作用和偏離中心的π…π相互作用得到最大化,並且滿足偶極作用,這種完美的組織方式能夠使所有的相互作用得到最大化,橋連雙萘之間的堆積能夠與脲基的氫鍵相互促進,在這些作用下能夠形成非常穩定的超分子聚合物。在非極性溶液中,這種效果得到放大,從而形成聚集度很高的超分子聚合物,這些聚合物進一步纏繞從而使這些溶劑形成凝膠。
另一方面,本發明提供了如上所述的相選擇性凝膠劑的製備方法,所述製備方法為:採用式II所示的異氰酸酯與式III所示的雙胺化合物反應得到所述相選擇性凝膠劑,反應式如下:
其中R為C8-C18(例如C8、C9、C10、C11、C12、C13、C14、C15、C16、C17、C18、)的直鏈烷基,優選C12直鏈烷基(即-(CH2)11CH3)。
優選地,所述式II所示的異氰酸酯與式III所示的雙胺化合物的摩爾比≥2:1,例如2:1、2.2:1、2.4:1、2.5:1、2.8:1、3:1、3.5:1、4:1等,優選(2-2.5):1。
優選地,所述反應在催化劑存在下進行,所述催化劑為N,N-二異丙基乙胺。在本發明中所述催化劑N,N-二異丙基乙胺除了可以催化反應外,還可以提高式III所示化合物在反應介質中的溶解性。
優選地,所述催化劑的用量為式II所示的異氰酸酯質量的3-8%,例如3%、3.5%、4%、4.5%、5%、5.5%、6%、6.5%、7%、7.5%或8%。
優選地,所述反應的溫度為20-30℃,例如20℃、21℃、23℃、25℃、27℃、29℃或30℃。
優選地,所述反應的時間為3-20h,例如3h、5h、8h、10h、12h、14h、16h、18h或20h。
優選地,所述反應的介質為二氯甲烷和/或三氯甲烷;
優選地,所述反應的介質為乾燥的二氯甲烷和/或三氯甲烷,即進行過除水乾燥處理的二氯甲烷和/或三氯甲烷。
第三方面,本發明提供了所述相選擇性凝膠劑在膠凝油類或有機溶劑中的應用。
優選地,所述油類為汽油、煤油、柴油或機油中的任意一種或至少兩種的組合。
優選地,所述有機溶劑為正己烷、環己烷、苯、甲苯或二甲苯中的任意一種或至少兩種的組合。
本發明的相選擇性凝膠劑可使油類或發生有機溶劑發生膠凝,具有油水分離能力,可用於治理油類以及有機溶劑洩漏。
相對於現有技術,本發明具有以下有益效果:
本發明通過將兩個脲基連接在橋連雙萘骨架上得到式I所示的凝膠劑,該凝膠劑可以選擇性地進入油相,兩個脲基可以促進橋連雙萘骨架之間的非共價相互作用,以發生聚集,使得油相發生膠凝,形成穩定的凝膠,可以使得油水分離,可用於油類以及有機溶劑洩漏的治理,並且該凝膠劑不溶於水,可以回收再利用,安全無汙染,在回收海上溢油和快速分離工業含油或有機溶劑廢水等領域具有很好的應用前景。
附圖說明
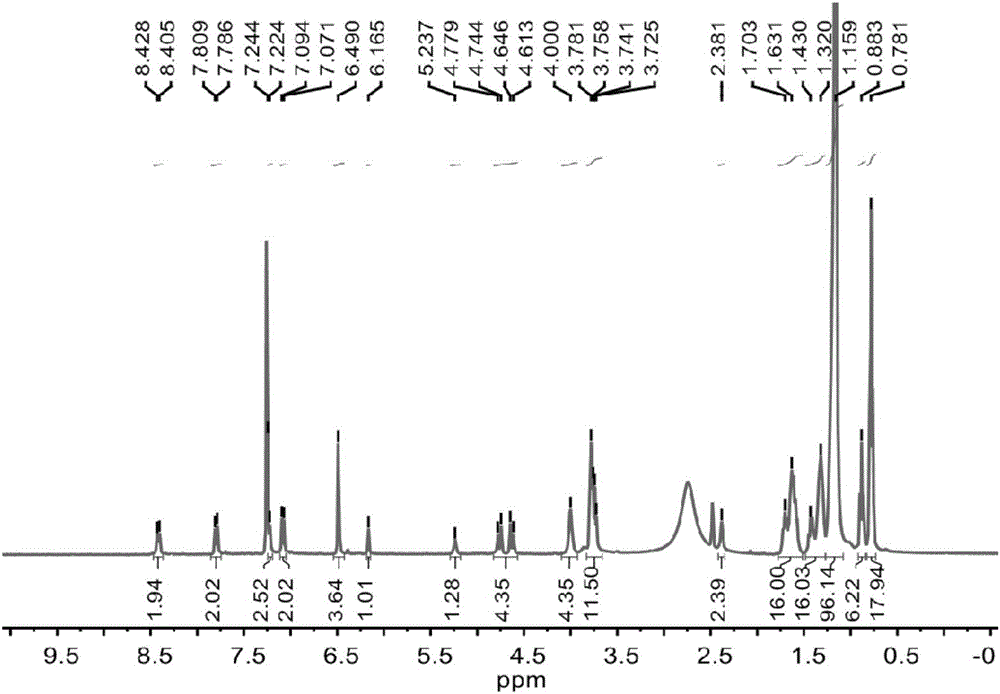
圖1為實施例1製備得到的凝膠劑的核磁氫譜圖。
圖2為實施例1製備得到的凝膠劑的核磁碳譜圖。
圖3為實施例1製備得到的凝膠劑的質譜圖。
圖4為本發明凝膠劑使汽油形成凝膠的過程示意圖。
圖5A為實施例1製備得到的凝膠劑的加入量為汽油質量的5.0%的情況下形成的凝膠在旋轉模式下,儲能模量(G')和損耗模量(G」)隨剪切速率的變化圖。
圖5B為實施例1製備得到的凝膠劑的加入量為汽油質量的5.0%的情況下形成的凝膠在震蕩模式下,儲能模量(G')和損耗模量(G」)隨剪切應力的變化圖。
圖6為在混合溶劑氘代氯仿和氘代DMSO(500:1)中不同濃度下的實施例1製備的凝膠劑的核磁氫譜圖;
圖7為本發明的凝膠劑在汽油中形成的凝膠製備成的幹凝膠的掃描電鏡圖,其標尺為5μm。
圖8為利用本發明的凝膠劑進行油水分離的過程示意圖。
具體實施方式
下面通過具體實施方式來進一步說明本發明的技術方案。本領域技術人員應該明了,所述實施例僅僅是幫助理解本發明,不應視為對本發明的具體限制。
實施例1
在本實施例中,通過以下反應式製備如下式I所示相選擇性凝膠劑:
其中R為-(CH2)11CH3。
製備過程為:
(1)參照文獻[Hong Yang,et al.Switchable Fluorescent Organogels and Mesomorphic Superstructure Based on Naphthalene Derivatives,Langmuir 2007,23,8224-8230]合成式II所示化合物,合成流程如下:
中間體S1的合成:在沒食子酸甲酯(18g,97.8mmol)與K2CO3(54g,390mmol)的DMF(90mL)懸浮液中加入溴代十二烷(75mL,300mmol),攪拌並加熱至70℃,在氬氣保護下反應過夜。然後將反應液倒入蒸餾水中(500mL)。過濾沉澱並用丙酮重結晶得到白色固體S1 60.6g,收率約為90%。
中間體S2的合成:將化合物S1(4g,6.8mmol)的乙醇溶液(30mL)與過量NaOH水溶液(10mL)混合,並在常溫下攪拌6小時。反應完後用1M的鹽酸調反應液pH至1。用二氯甲烷萃取有機相,並用無水Na2SO4乾燥,旋蒸得到白色固體S2 3.5g,收率約為90%。
式II化合物的合成:取中間體化合物S2(2.8g,4.1mmol)溶於過量的SOCl2(14mL)中,攪拌並加熱至回流,反應4小時。反應完後,旋蒸掉剩餘的SOCl2,得到醯氯。取過量NaN3(2g,30mmol)溶於H2O(30mL)。在冰浴下,將上述醯氯的THF(10mL)溶液逐滴加入NaN3水溶液中,常溫下攪拌並反應過夜。反應完後用二氯甲烷萃取有機相,並用飽和食鹽水洗滌,無水Na2SO4乾燥,旋蒸得到黃色固體。將此黃色固體在油泵下減壓乾燥後,溶於超幹的甲苯(40mL),氬氣保護,攪拌並加熱至回流,反應6小時。之後將溶劑旋幹,得到式II化合物。
(2)參照文獻[Zhenfeng He,et al.Imine Macrocycle with a Deep Cavity:Guest-Selected Formation of syn/anti Configuration and Guest-Controlled Reconfiguration(Supporting Information),Chemistry A European Journal,2015,21,3005-3012.]合成式III所示化合物,合成流程如下:
中間體化合物C1的合成:無水K2CO3(4.5g,32.5mmol)加入到溶有2,6-二羥基萘(4.0g,25.0mmol)的DMF(200mL)溶液中加熱攪拌1h後,通過恆壓滴液漏鬥將正溴丁烷(3.2mL,30.0mmol)緩慢加入到反應液中。加熱80℃反應7h。冷卻至室溫,向反應液中加入大量水並加入少量稀鹽酸(pH=7)攪拌,大量不溶固體析出。過濾並用大量水洗滌濾渣,將濾渣溶於200mL甲醇中,攪拌過濾,得到的濾液加入無水硫酸鈉乾燥過夜,過濾將得到的濾液蒸乾。得到的殘留物通過柱層析分離(石油醚/二氯甲烷=1/1,v/v),得到白色固體(1.6g,30%)。
中間體化合物C2的合成:化合物C1(4.0g,18.5mmol)溶於到三氟乙酸(TFA,25mL)和四氫呋喃(THF,100mL)的混合溶液中,通過恆壓滴液漏鬥將丙二縮醛(1.6mL,9.7mmol)緩慢加入到反應液中,室溫反應3h。向其中加入1mol/L NaOH溶液、飽和Na2HCO3溶液,調節pH=8。用二氯甲烷萃取有機相後,加入無水Na2SO4乾燥,旋蒸乾溶劑,殘留物通過柱層析分離(PE/DCM=5/1,v/v)得到白色固體C2(3.4g,80%)。
中間體化合物C3的合成:冰浴下,將化合物C2(1.5g,3.2mmol)溶於乾燥的二氯甲烷(30mL)中,邊攪拌邊緩慢加入1,1-二氯甲醚(7.5mL,13mmol)和四氯化鈦(2.2g,11.6mmol)。冰浴下攪拌反應1小時後升至室溫再繼續反應2.5h。反應完成後向反應液中加入冰水混合物,並加入大量飽和NaHCO3攪拌使其pH=8。用二氯甲烷萃取有機相,加入無水Na2SO4乾燥。過濾後旋蒸乾溶劑並通過柱層析分離(PE/DCM=1/1,v/v)得到黃色固體C3(1.4g,86%)。
中間體化合物C4的合成:化合物C3(0.6g,1.1mmol)溶於MeCN(20mL)與DCM(40mL)的混合溶液中,向其中逐步加入氨基甲酸叔丁酯(0.8g,6.9mmol)、三乙基矽烷(1.1mL,7.1mmol)和三氟乙酸(1mL,5.0mmol),室溫攪拌反應過夜。加入飽和NaHCO3攪拌使其pH=8,用二氯甲烷萃取有機相,加入無水Na2SO4乾燥。過濾後旋蒸乾溶劑並通過柱層析分離(PE/DCM=2/1,v/v)得到白色固體(0.6g,76%)。
式III化合物的合成:化合物C4(1.0g,1.9mmol)溶於三氟乙酸(15mL)與DCM(30mL)的混合溶液中室溫攪拌4小時。旋蒸乾溶劑後將剩餘固體進一步抽乾得到褐色固體粉末。將其溶於二氯甲烷中攪拌後過濾得到白色固體,加入二氯甲烷(30mL)和1M NaOH溶液(20mL)攪拌,固體逐漸溶解。萃取有機相,加入無水Na2SO4乾燥,過濾後旋蒸乾溶劑得到淡黃色固體(0.6g,71%)。
1H NMR(400MHz,CDCl3,300K):ppm=8.47(d,J=9.2,2H),7.81(d,J=9.2,2H),7.32(d,J=9.2,2H),7.19(d,J=9.2,2H),6.25(s,1H),5.29(s,1H),4.18(s,4H),4.15-4.02(m,4H),2.47-2.35(m,2H),1.88-1.79(m,4H),1.62-1.47(m,4H),1.00(t,J=7.4Hz,6H);13C NMR(100MHz,CDCl3,300K):ppm=152.26,148.45,128.65,126.77,125.62,123.11,123.01,119.38,119.05,114.44,91.38,69.01,36.37,31.73,27.06,23.03,19.43,13.92;ESI-TOF-HRMS:m/z calcd for[M+Na]+C33H38N2O4Na 549.2724;found 549.2725。
(3)取式III化合物(800mg,1.5mmol)和N,N-二異丙基乙胺(0.2mL)溶於乾燥的二氯甲烷(20mL)中。常溫下,通過恆壓滴液漏鬥將式II化合物(約2.4g,3.6mmol)的乾燥二氯甲烷(5mL)溶液滴加入上述溶液中,滴加時間30分鐘,將得到的混合物再在常溫下攪拌5個小時。反應完成後,過濾沉澱並依次用二氯甲烷和甲醇洗滌,得到白色固體2.5g,收率約為89%。
對本實施例製備得到的白色固體產物進行核磁氫譜、核磁碳譜和質譜表徵,結果如圖1-3所示,其數據結果如下:
1H NMR(400MHz,CDCl3:DMSO-d6=99:1,25℃):δ[ppm]=8.42(d,J=9.3Hz,2H),7.80(d,J=9.3Hz,2H),7.23(d,J=8.1Hz,2H),7.08(d,J=9.2Hz,2H),6.49(s,4H),6.16(s,1H),5.24(s,1H),4.70(dd,J=52.7,13.4Hz,4H),4.07-3.90(m,4H),3.87-3.65(m,12H),2.38(s,2H),1.81-1.52(m,16H),1.50–1.26(m,16H),1.16(s,96H),0.93-0.84(m,6H),0.82-0.70(m,18H).13C NMR(126MHz,CDCl3:DMSO-d6=10:1,25℃):δ[ppm]=155.34,152.61,152.46,148.22,135.40,132.20,128.67,126.01,123.62,120.55,119.03,118.65,96.85,90.81,72.86,69.09,68.30,33.48,31.39,31.13,29.78,29.22,29.20,29.17,29.12,28.91,28.85,28.82,25.65,25.61,22.21,22.16,18.80,13.67,13.47.ESI-TOF-HRMS:m/z calcd for[M+H]+C119H193O12N4+,1870.4592;found 1870.4610(error=-0.9623ppm)。
該結果證明製備得到的白色固體產物具有本實施例式I所示結構。
實施例2
在本實施例中,通過與實施例1相同的方法製備得到式II化合物和式III化合物;通過以下方法製備得到式I化合物:
取式III化合物3(800mg,1.5mmol)和N,N-二異丙基乙胺(0.07mL)溶於乾燥的二氯甲烷(20mL)中。常溫下,通過恆壓滴液漏鬥將式II化合物(約2g,3.0mmol)的乾燥二氯甲烷(5mL)溶液滴加入上述溶液中,滴加時間30分鐘,將得到的混合物再在30℃下攪拌12個小時。反應完成後,過濾沉澱並依次用二氯甲烷和甲醇洗滌,得到白色固體2.3g,收率約為82%。
1H NMR(400MHz,CDCl3:DMSO-d6=99:1,25℃):δ[ppm]=8.42(d,J=9.3Hz,2H),7.80(d,J=9.3Hz,2H),7.23(d,J=8.1Hz,2H),7.08(d,J=9.2Hz,2H),6.49(s,4H),6.16(s,1H),5.24(s,1H),4.70(dd,J=52.7,13.4Hz,4H),4.07-3.90(m,4H),3.87-3.65(m,12H),2.38(s,2H),1.81-1.52(m,16H),1.50–1.26(m,16H),1.16(s,96H),0.93-0.84(m,6H),0.82-0.70(m,18H).13C NMR(126MHz,CDCl3:DMSO-d6=10:1,25℃):δ[ppm]=155.34,152.61,152.46,148.22,135.40,132.20,128.67,126.01,123.62,120.55,119.03,118.65,96.85,90.81,72.86,69.09,68.30,33.48,31.39,31.13,29.78,29.22,29.20,29.17,29.12,28.91,28.85,28.82,25.65,25.61,22.21,22.16,18.80,13.67,13.47.ESI-TOF-HRMS:m/z calcd for[M+H]+C119H193O12N4+,1870.4592;found 1870.4613。
實施例3
在本實施例中,通過與實施例1相同的方法製備得到式II化合物和式III化合物;通過以下方法製備得到式I化合物:
取式III化合物3(800mg,1.5mmol)和N,N-二異丙基乙胺(0.25mL)溶於乾燥的二氯甲烷(20mL)中。25℃常溫下,通過恆壓滴液漏鬥將式II化合物(約2.5g,3.75mmol)的乾燥二氯甲烷(5mL)溶液滴加入上述溶液中,滴加時間30分鐘,將得到的混合物再在25℃常溫下攪拌12個小時。反應完成後,過濾沉澱並依次用二氯甲烷和甲醇洗滌,得到白色固體2.4g,收率約為85%。
1H NMR(400MHz,CDCl3:DMSO-d6=99:1,25℃):δ[ppm]=8.42(d,J=9.3Hz,2H),7.80(d,J=9.3Hz,2H),7.23(d,J=8.1Hz,2H),7.08(d,J=9.2Hz,2H),6.49(s,4H),6.16(s,1H),5.24(s,1H),4.70(dd,J=52.7,13.4Hz,4H),4.07-3.90(m,4H),3.87-3.65(m,12H),2.38(s,2H),1.81-1.52(m,16H),1.50–1.26(m,16H),1.16(s,96H),0.93-0.84(m,6H),0.82-0.70(m,18H).13C NMR(126MHz,CDCl3:DMSO-d6=10:1,25℃):δ[ppm]=155.34,152.61,152.46,148.22,135.40,132.20,128.67,126.01,123.62,120.55,119.03,118.65,96.85,90.81,72.86,69.09,68.30,33.48,31.39,31.13,29.78,29.22,29.20,29.17,29.12,28.91,28.85,28.82,25.65,25.61,22.21,22.16,18.80,13.67,13.47.ESI-TOF-HRMS:m/z calcd for[M+H]+C119H193O12N4+,1870.4592;found 1870.4615。
實施例4
在本實施例中,通過以下反應式製備如下式I所示相選擇性凝膠劑:
其中R為-(CH2)7CH3。
首先通過與實施例(1)中步驟(1)相同的方法來合成式II化合物:在合成過程中將實施例1步驟(1)中溴代十二烷替換成溴代正辛烷,合成得到式II化合物,經核磁氫譜驗證了式II化合物結構的正確性。
以與實施例1中步驟(2)相同的方法合成得到式III化合物;
通過以下步驟(3)製備得到式I所示凝膠劑,即取式III化合物(800mg,1.5mmol)和N,N-二異丙基乙胺(0.1mL)溶於乾燥的二氯甲烷(20mL)中。常溫下,通過恆壓滴液漏鬥將式II化合物(約1.6g,3.3mmol)的乾燥二氯甲烷(5mL)溶液滴加入上述溶液中,滴加時間30分鐘,將得到的混合物再在20℃下攪拌20個小時。反應完成後,過濾沉澱並依次用二氯甲烷和甲醇洗滌,得到白色固體2.0g,收率約為87%。質譜測試數據如下:ESI-TOF-HRMS:m/z calcd for[M+H]+C95H145O12N4+,1535.1795;found 1535.1788。
實施例5
在本實施例中,通過以下反應式製備如下式I所示相選擇性凝膠劑:
其中R為-(CH2)17CH3。
首先通過與實施例(1)中步驟(1)相同的方法來合成式II化合物:在合成過程中將實施例1步驟(1)中溴代十二烷替換成溴代正十八烷,合成得到式II化合物,經核磁氫譜驗證了式II化合物結構的正確性。
以與實施例1中步驟(2)相同的方法合成得到式III化合物;
通過以下步驟(3)製備得到式I所示凝膠劑,即取式III化合物(800mg,1.5mmol)和N,N-二異丙基乙胺(0.16mL)溶於乾燥的二氯甲烷(20mL)中。常溫下,通過恆壓滴液漏鬥將式II化合物(約3.2g,3.45mmol)的乾燥二氯甲烷(5mL)溶液滴加入上述溶液中,滴加時間30分鐘,將得到的混合物再在25℃下攪拌15個小時。反應完成後,過濾沉澱並依次用二氯甲烷和甲醇洗滌,得到白色固體1.74g,收率約為85%。質譜測試數據如下:ESI-TOF-HRMS:m/z calcd for[M+H]+C155H265O12N4+,2376.7743;found 2376.7745。
實施例6
將實施例1製備得到的式I所示凝膠劑粉末放入裝有汽油的小瓶中,其用量為小瓶中汽油質量的1.0%,在室溫下靜置,大約3小時之後,式I所示凝膠劑粉末擴散入汽油中形成透明澄清的溶液,還未形成凝膠。然而,再過3小時,汽油溶液轉變成了凝膠,該過程示意圖如圖4所示。在其他的油類(例如煤油、柴油或機油)以及有機溶劑(例如正己烷、環己烷、苯、甲苯或二甲苯)中也能看到類似的現象,說明該凝膠劑在這些溶液中有很好的溶解性,能夠促使這些油類和有機溶劑發生膠凝,測試了在油類和一些有機溶劑中的臨界成膠濃度(CGC)和以及在測定濃度下的成膠溫度(Tgel),如表1所示。
表1
由表1的結果可以表明,式I所示凝膠劑可以使得油類和有機溶劑發生膠凝,是很好的凝膠劑,形成的凝膠比較穩定。
在該實施例中,對在加入實施例1製備得到的式I所示凝膠劑為汽油質量的5.0%的情況下形成的凝膠的流變性質進行考察,結果如圖5A和圖5B所示,在旋轉模式下(圖5A),儲能模量(G')一直高於損耗模量(G」),並且這兩種模量都隨頻率的變化而變化,證明是一種較強的凝膠。在震蕩模式下(圖5B),壓力產量(大約100pa)和應力幅(σ0)表明該凝膠強度足以承擔自己的重量,說明形成的凝膠具有較好的機械強度,穩定性較好。
為了驗證式I所示相選擇性凝膠劑分子發生膠凝的時分子中的相互作用,在不同的濃度下測試膠凝劑在混合溶劑氘代氯仿和氘代DMSO(500:1)中的核磁氫譜(1H NMR),如圖6所示,可以看出,隨著膠凝劑在混合溶劑中濃度的增加,相對的尖銳的信號峰逐漸變寬,表明膠凝劑分子發生了聚集,同時,位於橋連雙萘芳香區的質子峰向高場發生位移,表明形成了「從背後擁抱」的類似於「超分子聚合物」的結構,從而發生了屏蔽效應,因此,具有膠凝劑特徵。
將式I所示凝膠劑在汽油中形成的凝膠製備成幹凝膠,利用掃描電鏡(FESEM,ZEISS Merlin)對其進行表徵,結果如圖7所示,從圖中可以看出,形成的幹凝膠存在糾纏的網絡,說明由式I所示凝膠劑形成的一維纖維進一步纏繞從而形成凝膠。分子模擬(即利用Spartan』14PM6級理論進行計算)表明橋連雙萘之間的堆積確實能夠與脲基的氫鍵相互促進。在這些作用下能夠形成非常穩定的超分子聚合物。在非極性溶液中,這種效果得到放大,從而形成聚集度很高的超分子聚合物。這些聚合物進一步纏繞從而使這些溶劑形成凝膠。
同樣,實施例2-5製備得到的凝膠劑同樣能夠使得油類(例如汽油、煤油、柴油或機油)以及有機溶劑(例如正己烷、環己烷、苯、甲苯或二甲苯)發生膠凝。
實施例7
在本實施例中,將實施例1-5製備得到的凝膠劑用於油水分離(實施例1-5製備得到的凝膠劑不溶於水並且形成沉澱),其過程示意圖如圖8所示,即在燒杯中將10mL水和0.5mL汽油的混合得到油水混合物,汽油分散漂浮在水面上,將凝膠劑粉末(圖中以1a表示)加入至燒杯中,加入量為汽油質量的2.0wt%,為了促進凝膠過程,將水相進行攪拌,6小時後汽油形成了凝膠,汽油收縮成一大塊的凝膠,用勺子很容易就能從水中勺出。通過蒸餾能夠收集回收的凝膠中的汽油並且凝膠劑能夠再次循環利用。因此,雖然這種凝膠劑可能成本較高,但它是能夠回收利用的。因此,本發明製備的凝膠劑可以用於治理油類洩漏。
申請人聲明,本發明通過上述實施例來說明本發明的相選擇性凝膠劑及其製備方法和應用,但本發明並不局限於上述實施例,即不意味著本發明必須依賴上述實施例才能實施。所屬技術領域的技術人員應該明了,對本發明的任何改進,對本發明所選用原料的等效替換及輔助成分的添加、具體方式的選擇等,均落在本發明的保護範圍和公開範圍之內。