一種V離子摻雜BiNb5O14光催化劑及其製備方法和應用與流程
2023-04-30 00:45:56 5
本本發明涉及一種無機光催化劑材料的製備方法及其應用,特別涉及用於降解有機汙染物的v離子摻雜binb5o14光催化劑及其製備方法和應用,屬於無機光催化材料領域。
背景技術:
目前,隨著全球工業化進程的不斷發展與進步,人們的生活水平不斷提高,全球經濟也不斷發展。但是同時環境與能源問題也越發嚴重。如今,環境問題已成為人類生存和社會可持續發展所面臨的最大挑戰。近年來,水汙染已經成為人們的憂患,尤其是染料汙水問題尤為突出。如何解決染料汙水問題已經成為主要研究對象之一。怎樣在低能或無能耗的條件下高效無殘留的去除水中染料成為研究者們的研究熱點。
光催化材料是指一種在光的照射下,自身不起變化,可以促進化學反應的物質。光催化材料可以有效降解許多結構穩定的有機汙染物,已成為國內外重視的汙染治理技術之一。光催化技術不僅能夠直接利用光的照射降解有機汙染物,也可以分解水獲得清潔能源。目前光催化技術已經成為汙染物處理與分解水制氫能源等領域研究方向和可能廣泛應用的技術。近年來,開發能夠在光照下快速高效降解有機染料的光催化劑已經成為研究者們的研究熱點。
中國專利cn201410687080.2報導了一種大比表面積鈮酸鹽光催化劑的製備方法及應用,同時中國專利cn201310252473.6報導了可見光響應的光催化劑bacuv2o7及其製備方法。以上兩個專利分別報導了鈮酸鹽和釩酸鹽光催化材料,為光催化材料的開發提供了新方向。因此在這些報導的基礎上,我們研究了一種v離子摻雜binb5o14光催化劑,發現該類化合物具有優異的光催化性能,而且目前尚無報導。
技術實現要素:
針對上述現有技術存在的問題,本發明提供v離子摻雜binb5o14光催化劑及其製備方法和應用,製備方法簡單、光催化效率高、應用前景廣闊。
一種v離子摻雜binb5o14光催化材料,它的化學式為binb5-5xv5xo14,其中x為v5+摻雜的摩爾百分數,0.0001≤x≤0.15。
上述製備的樣品為單一的純相材料物。
一種v離子摻雜binb5o14光催化材料的製備方法,包括如下步驟:
(1)按化學式binb5-5xv5xo14中各元素的化學計量比,首先稱取含有鉍離子bi3+的化合物,溶於適量的稀硝酸溶液中,加熱攪拌,直至完全溶解,再加入適量的絡合劑,攪拌溶解得到a溶液;稱取含有鈮離子nb5+的化合物,溶於適量的稀硝酸溶液中,加熱攪拌,直至完全溶解,再加入適量的絡合劑,攪拌溶解得到b溶液;稱取含有釩離子v5+的化合物,溶於適量的稀硝酸溶液中,加熱攪拌,直至完全溶解,再加入適量的絡合劑,攪拌溶解得到c溶液,最後,將a、b、c溶液混合,繼續攪拌1-5小時得到混合均勻溶液;
(2)將上述混合物溶液放置在烘箱中,溫度為50℃-100℃,時間為12小時,得到蓬鬆的前驅體;
(3)自然冷卻後,取出前驅體,在空氣氣氛中煅燒,煅燒溫度為750~1400℃,煅燒時間為2~16小時,自然冷卻後,研磨均勻即得到一種v離子摻雜binb5o14光催化粉末材料。
所述的含鉍離子bi3+的化合物為氧化鉍bi2o3、硝酸鉍bi(no3)3•5h2o、氫氧化鉍bi(oh)3和氯化鉍bicl3中的一種;所述的含有鈮離子nb5+的化合物為五氧化二鈮nb2o5、氫氧化鈮nb(oh)5和氯化鈮nbcl5中的一種;所述的含有釩離子v5+的化合物為五氧化二釩v2o5、偏釩酸銨nh4vo3中的一種。
步驟(3)所述的煅燒溫度為800~1350℃,煅燒時間為3~15小時。
上述所述的v離子摻雜binb5o14材料能夠作為無機光催化材料,光催化效率高、應用前景廣闊。
與現有技術方案相比,本發明技術方案優點在於:
(1)製備的v離子摻雜binb5o14光催化劑的原材料來源很廣泛且價格廉,製備方法簡單,生產成本低。
(2)製備的v離子摻雜binb5o14光催化劑物相較純,且分布均勻。
(3)製備的v離子摻雜binb5o14光催化劑的光催化活性非常好,120分鐘光催化降解亞甲基藍的降解率可以達到93%,能夠高效地光催化降解亞甲基藍。
(4)本發明無廢氣廢液排放,v離子摻雜binb5o14光催化劑是一種綠色安全的無機光催化材料。
附圖說明
圖1為本發明實施例1所製得的binb4.9995v0.0005o14樣品的x射線粉末衍射圖譜;
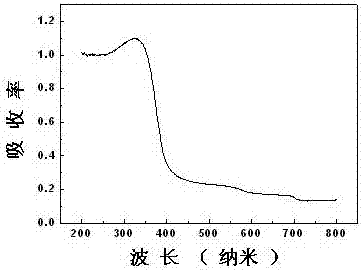
圖2為本發明實施例1所製得的binb4.9995v0.0005o14樣品的紫外可見吸收光圖譜;
圖3為本發明實施例1所製得的binb4.9995v0.0005o14樣品的sem圖;
圖4為本發明實施例1所製得的binb4.9995v0.0005o14樣品在光照時對有機染料亞甲基藍的降解曲線;
圖5為本發明實施例4所製得的binb4.75v0.25o14樣品的x射線粉末衍射圖譜;
圖6為本發明實施例4所製得的binb4.75v0.25o14樣品的紫外可見吸收光圖譜;
圖7為本發明實施例4所製得的binb4.75v0.25o14樣品的sem圖;
圖8為本發明實施例4所製得的binb4.75v0.25o14樣品在光照時對有機染料亞甲基藍的降解曲線;
圖9為本發明實施例6所製得的binb4.25v0.75o14樣品的x射線粉末衍射圖譜;
圖10為本發明實施例6所製得的binb4.25v0.75o14樣品的紫外可見吸收光圖譜。
具體實施方式
下面結合附圖和實施例對本發明技術方案作進一步描述。
1、為了得到本發明中所使用的複合氧化物,首先使用固相合成法製備粉末,即把原料按照目標組成化學計量比進行混合,再在常壓下於空氣氣氛中合成。
2、為了能夠有效利用光,本發明中的光催化劑的尺寸最好在微米級別,甚至是納米粒子,且比表面積較大。用固相合成法製備的氧化物粉末,其粒子較大而表面積較小,但是可以通過改用化學溶液法製備光催化劑使粒子直徑變小。
3、光催化降解亞甲基藍活性評價採用自製光催化反應裝置,光源燈為500瓦圓柱形形氙燈,反應槽使用硼矽酸玻璃製成的圓柱形光催化反應儀器,將光源燈插入到反應槽中,並通入冷凝水降溫,反應時溫度為室溫。催化劑用量100毫克,溶液體積250毫升,亞甲基藍的濃度為10毫克/升。催化劑置於反應液中,催化時間設定為240分鐘,打開冷凝水後開始光照,光照後每隔一段時間取一次樣,離心,取其上清液,用紫外-可見分光光度計在波長664-666納米處測定亞甲基藍溶液的吸光度。根據朗伯-比爾定律,溶液的吸光度與濃度成正比,因此可用吸光度代替濃度計算去除率,以此為亞甲基藍溶液的去除率。計算公式:降解率=(1-c/c0)×100%=(1-a/a0)×100%,其中c0、c分別為光催化降解前後的濃度,a0、a分別是降解前後的吸光度值。
實施例1:
根據化學式binb4.9995v0.0005o14,分別稱取氧化鉍bi2o3:2.3298克,五氧化二鈮nb2o5:6.6445克,五氧化二釩v2o5:0.0005克,分別加入稀硝酸溶液中攪拌直至完全溶解再加入適量的檸檬酸,磁力攪拌一段時間後分別得到a、b、c溶液,將三種溶液混合,繼續攪拌一段時間。最終將得到的混合溶液放置烘箱中,設定溫度為80℃,烘12小時後,自然冷卻,取出蓬鬆的前驅體,在空氣氣氛中煅燒,煅燒溫度為800℃,煅燒時間為3小時,冷卻研磨即得到binb4.9995v0.0005o14光催化劑。
參見附圖1,它是按本實施例技術方案所製備樣品的x射線粉末衍射圖譜,xrd測試結果顯示,所製備的樣品為單一的binb5o14晶體結構,且結晶度較好;
參見附圖2,它是按本實施例技術方案所製備樣品的紫外可見吸收光譜,從圖中可以看出,該樣品在紫外光區域具有很強的吸收,說明此樣品是紫外光響應的光催化劑;
參見附圖3,它是按本實施例技術方案所製備樣品的sem(掃描電子顯微鏡)圖譜,從圖中可以看出樣品的形貌;
參見附圖4,它是按本實施例技術方案所製備樣品對有機染料亞甲基藍的降解曲線。從圖中可以看出,該樣品光催化降解亞甲基藍的降解率120分鐘可以達到80%,說明製備出的binb4.9995v0.0005o14材料具有較好的的光催化活性。
實施例2:
根據化學式binb4.995v0.005o14,分別稱取硝酸鉍bi(no3)3•5h2o:4.8510克,氫氧化鈮nb(oh)5:8.8864克,偏釩酸銨nh4vo3:0.0058克,分別加入稀硝酸溶液中攪拌直至完全溶解再加入適量的檸檬酸,磁力攪拌一段時間後分別得到a、b、c溶液,將三種溶液混合,繼續攪拌一段時間。最終將得到的混合溶液放置烘箱中,設定溫度為80℃,烘12小時後,自然冷卻,取出蓬鬆的前驅體,在空氣氣氛中煅燒,煅燒溫度為900℃,煅燒時間為6小時,冷卻研磨即得到binb4.995v0.005o14光催化劑。
其主要的晶體結構、紫外可見吸收光譜、sem圖譜、對亞甲基藍的降解曲線與實施例1相似。
實施例3:
根據化學式binb4.95v0.05o14,分別稱取氯化鈮nbcl5:3.1534克,氯化鈮nbcl5:13.3743克,五氧化二釩v2o5:0.0455克,分別加入稀硝酸溶液中攪拌直至完全溶解再加入適量的檸檬酸,磁力攪拌一段時間後分別得到a、b、c溶液,將三種溶液混合,繼續攪拌一段時間。最終將得到的混合溶液放置烘箱中,設定溫度為80℃,烘12小時後,自然冷卻,取出蓬鬆的前驅體,在空氣氣氛中煅燒,煅燒溫度為1000℃,煅燒時間為8小時,冷卻研磨即得到binb4.95v0.05o14光催化劑。
其主要的晶體結構、紫外可見吸收光譜、sem圖譜、對亞甲基藍的降解曲線與實施例1相似。
實施例4:
根據化學式binb4.75v0.25o14,分別稱取氫氧化鉍bi(oh)3:2.6000克,氫氧化鈮nb(oh)5:8.4504克,偏釩酸銨nh4vo3:0.2924克,分別加入稀硝酸溶液中攪拌直至完全溶解再加入適量的檸檬酸,磁力攪拌一段時間後分別得到a、b、c溶液,將三種溶液混合,繼續攪拌一段時間。最終將得到的混合溶液放置烘箱中,設定溫度為80℃,烘12小時後,自然冷卻,取出蓬鬆的前驅體,在空氣氣氛中煅燒,煅燒溫度為1100℃,煅燒時間為10小時,冷卻研磨即得到binb4.75v0.25o14光催化劑。
參見附圖5,它是按本實施例技術方案所製備樣品的x射線粉末衍射圖譜,xrd測試結果顯示,所製備的樣品為單一的binb5o14晶體結構,說明v離子的引入沒有影響晶體的結構;
參見附圖6,它是按本實施例技術方案所製備樣品的紫外可見吸收光譜,從圖中可以看出,該樣品在紫外光區域具有很強的吸收;
參見附圖7,它是按本實施例技術方案所製備樣品的sem(掃描電子顯微鏡)圖譜,從圖中可以看出所得樣品顆粒分散均勻;
參見附圖8,它是按本實施例技術方案所製備樣品對有機染料亞甲基藍的降解曲線。從圖中可以看出,該樣品光催化降解亞甲基藍的降解率120分鐘可以達到93%,說明製備出的binb4.75v0.25o14材料具有非常好的的光催化活性。
實施例5:
根據化學式binb4.5v0.5o14,分別稱取氧化鉍bi2o3:2.3298克,五氧化二鈮nb2o5:5.9807克,五氧化二釩v2o5:0.4550克,分別加入稀硝酸溶液中攪拌直至完全溶解再加入適量的檸檬酸,磁力攪拌一段時間後分別得到a、b、c溶液,將三種溶液混合,繼續攪拌一段時間。最終將得到的混合溶液放置烘箱中,設定溫度為80℃,烘12小時後,自然冷卻,取出蓬鬆的前驅體,在空氣氣氛中煅燒,煅燒溫度為1200℃,煅燒時間為12小時,冷卻研磨即得到binb4.75v0.25o14光催化劑。
其主要的晶體結構、紫外可見吸收光譜、sem圖譜、對亞甲基藍的降解曲線與實施例4相似。
實施例6:
根據化學式binb4.25v0.75o14,分別稱取硝酸鉍bi(no3)3•5h2o:4.8510克,五氧化二鈮nb2o5:5.6485克,偏釩酸銨nh4vo3:0.8774克,分別加入稀硝酸溶液中攪拌直至完全溶解再加入適量的檸檬酸,磁力攪拌一段時間後分別得到a、b、c溶液,將三種溶液混合,繼續攪拌一段時間。最終將得到的混合溶液放置烘箱中,設定溫度為80℃,烘12小時後,自然冷卻,取出蓬鬆的前驅體,在空氣氣氛中煅燒,煅燒溫度為1350℃,煅燒時間為15小時,冷卻研磨即得到binb4.75v0.25o14光催化劑。
參見附圖9,它是按本實施例技術方案所製備樣品的x射線粉末衍射圖譜,xrd測試結果顯示,所製備的樣品為單一的binb5o14晶體結構,且結晶度非常好,說明v離子的引入沒有影響晶體的結構;
參見附圖10,它是按本實施例技術方案所製備樣品的紫外可見吸收光譜,從圖中可以看出,該樣品在紫外光區域具有很強的吸收,說明此樣品是一種紫外光響應的光催化劑材料;
其主要的sem圖譜、對亞甲基藍的降解曲線與實施例4相似。