一種中間體化合物以及丙硫菌唑的合成方法與流程
2023-04-26 18:38:01 2
本發明屬於有機合成領域,具體涉及一種中間體化合物以及丙硫菌唑的合成方法。
背景技術:
丙硫菌唑是一種脫甲基化抑制劑(DMIs),其作用機理是抑制真菌中甾醇的前體—羊毛甾醇l4-位脫甲基化反應。丙硫菌唑不僅具有很好的內吸活性,優異的保護、治療和剷除活性,且持效期長。大量的田間藥效試驗結果表明丙硫菌唑對作物不僅具有良好的安全性,防病治病效果好,而且增產明顯,同三唑類殺菌劑相比,丙硫菌唑具有更廣譜的殺菌活性。
丙硫菌唑目前主要用於防治禾穀類作物如小麥、大麥、油菜、花生、水稻和豆類作物等眾多病害。丙硫菌唑幾乎對所有麥類病害都有很好的防治效果,如小麥和大麥的白粉病、紋枯病、枯萎病、葉斑病、銹病、菌核病、網斑病、雲紋病等。丙硫菌唑還能防治油菜和花生的土傳病害,如菌核病,以及主要葉面病害,如灰黴病、黑斑病、褐斑病、黑脛病和銹病等。
根據硫原子的來源不同,丙硫菌唑的製備策略可分為兩大類。第一類製備丙硫菌唑的策略是以羥基三唑化合物為關鍵中間體,與硫磺反應得丙硫菌唑(US4913727)。硫磺在這類反應中作為丙硫菌唑化合物硫原子的來源。該方法的關鍵中間體可以氯化物(US4913727)或環氧化合物(US5146001)為起始原料和三氮唑經取代反應製得。此取代反應會同時產生相當量的區域異構體,需要通過精製去除,導致收率欠佳(51~53%)。關鍵中間體也可以氯代酮為原料和三氮唑反應,再和格氏試劑反應製得,此方法同樣存在區域選擇性問題。
美國專利US5789430公開了以化合物和硫磺直接反應製備丙硫菌唑的方法。該反應以N-甲基吡咯酮為溶劑在200℃下反應44小時得丙硫菌唑,收率為20%。US5789430同時也公開了以化合物和硫磺反應製備丙硫菌唑的改進方法。該改進方法是將化合物在THF溶劑裡先用n-BuLi拔氫,再和硫磺反應,所得丙硫菌唑的收率大大提高(93%),但該技術方案需要無水無氧和超低溫反應設備和條件,同時需要使用大於兩當量的高危險性的n-BuLi試劑,使該技術方案成本高,且操作不安全,不利於工業化生產。
另外,該技術方案同時也受自身化學區域選擇性的困擾,例如:(1)在關鍵中間體用n-BuLi拔氫過程中若控制不當將導致區域異構雜質11的產生;(2)在製備關鍵中間體時如果區域異構體不被完全分離提純乾淨,將導致區域異構雜質的產生。這些高要求的分離提純不僅產生大量三廢,同時大大增加成本。
US2013005985公開了一種使用格氏試劑如i-PrMgCl代替n-BuLi對化合物進行拔氫後再硫化製備丙硫菌唑的方法。該方法解決了使用n-BuLi試劑危險性問題,但該技術方案還是需要無水無氧和超低溫反應設備和條件,同時需要使用大於兩當量的格氏試劑,並且產率大幅下降(從n-BuLi的93%下降到68%)。
技術實現要素:
本部分的目的在於概述本發明的實施例的一些方面以及簡要介紹一些較佳實施例。在本部分以及本申請的說明書摘要和發明名稱中可能會做些簡化或省略以避免使本部分、說明書摘要和發明名稱的目的模糊,而這種簡化或省略不能用於限制本發明的範圍。
鑑於上述和/或現有合成丙硫菌唑的技術空白,提出了本發明。
因此,本發明其中的一個目的是提供一種中間體化合物,該中間體化合物能夠用於丙硫菌唑的合成。
為解決上述技術問題,本發明提供了如下技術方案:一種中間體化合物,其化學結構式為:
本發明其中的另一個目的是解決現有技術中丙硫菌唑合成方法的不足,提高每一步的轉化率,減少副產物,簡化反應條件,降低能耗,縮短反應時間,提高原子利用率,減少三廢。
為解決上述技術問題,本發明提供了如下技術方案:一種丙硫菌唑的合成方法,包括,以1,2,4-三氮唑為原料經硫氧化得到巰基-1,2,4-三氮唑;巰基-1,2,4-三氮唑經氧化形成5,5』-二硫基-雙(1,2,4-三氮唑);與2-(1-氯環丙基)-3-氯-1-(2-氯苯基)-2-丙醇發生取代反應得到如權利要求1所述中間體化合物;經還原得到目標產物丙硫菌唑。
作為本發明所述丙硫菌唑的合成方法的一種優選方案,其中:所述以1,2,4-三氮唑為原料經硫氧化得到巰基-1,2,4-三氮唑,包括,將1,2,4-三氮唑溶於有機溶劑中,加入同1,2,4-三氮唑摩爾比為1:3~1:6的硫,加熱至100~180℃,反應2~8h,,冷卻至室溫,過濾,濾液用飽和氯化鈉洗滌,經乙酸乙酯萃取,分出有機相,乾燥,蒸除乙酸乙酯得巰基-1,2,4-三氮唑;其中,有機溶劑為DMF或甲苯或DMSO中一種或多種;其中,乾燥採用固體乾燥劑為無水硫酸鎂、無水氯化鈣、無水硫酸鈉、無水硫酸鈣或活性氧化鋁中的一種或幾種。
作為本發明所述丙硫菌唑的合成方法的一種優選方案,其中:所述加熱至100~180℃,其溫度優選為110~160℃。
作為本發明所述丙硫菌唑的合成方法的一種優選方案,其中:所述反應2~8h,其反應時間優選為3~6h。
作為本發明所述丙硫菌唑的合成方法的一種優選方案,其中:所述巰基-1,2,4-三氮唑經氧化形成5,5』-二硫基-雙(1,2,4-三氮唑),包括,將巰基-1,2,4-三氮唑溶於DCM中,加入吡啶,控制溫度為-2℃~6℃,攪拌,加入苯磺醯氯,反應後去除DCM,剩餘物加入水和乙酸乙酯,反應後過濾,並用水和乙酸乙酯洗滌,將過濾得到的固體產物乾燥,得到固體化合物(Ⅴ)即5,5』-二硫基-雙(1,2,4-三氮唑);其中,乾燥採用固體乾燥劑為無水硫酸鎂、無水氯化鈣、無水硫酸鈉、無水硫酸鈣或活性氧化鋁中的一種或幾種。
作為本發明所述丙硫菌唑的合成方法的一種優選方案,其中:所述反應溫度優選為0℃~4℃。
作為本發明所述丙硫菌唑的合成方法的一種優選方案,其中:所述與2-(1-氯環丙基)-3-氯-1-(2-氯苯基)-2-丙醇發生取代反應,包括,將所述固體化合物(Ⅴ)和有機溶劑、碳酸鉀攪拌混合,加熱至20℃~100℃,加入2-(1-氯環丙基)-3-氯-1-(2-氯苯基)-2-丙醇中間體化合物,反應後冷卻至室溫,抽濾,濾液用飽和食鹽水洗滌,乙酸乙酯萃取,有機相用飽和食鹽水洗滌,分出有機相,乾燥,旋蒸得到化合物(Ⅵ);其中,有機溶劑為甲醇、乙醇、異丙醇、乙腈、丙酮或DMF中的一種或幾種;其中,固體化合物(V)同碳酸鉀摩爾比為1:2~1:4;其中,固體化合物(V)同2-(1-氯環丙基)-3-氯-1-(2-氯苯基)-2-丙醇化合物(Ⅱ)摩爾比為1:2~1:5;其中,乾燥採用固體乾燥劑為無水硫酸鎂、無水氯化鈣、無水硫酸鈉、無水硫酸鈣或活性氧化鋁中的一種或幾種。
作為本發明所述丙硫菌唑的合成方法的一種優選方案,其中:所述固體乾燥劑,優選為無水硫酸鈉。
作為本發明所述丙硫菌唑的合成方法的一種優選方案,其中:所述加熱至20℃~100℃,優選40℃~80℃。
作為本發明所述丙硫菌唑的合成方法的一種優選方案,其中:所述有機溶劑為,優選為DMF。
作為本發明所述丙硫菌唑的合成方法的一種優選方案,其中:所述經還原得到目標產物丙硫菌唑,包括,將化合物(Ⅵ)溶於有機溶劑中,加入還原劑,反應溫度20℃-60℃條件下攪拌反應後,重結晶,析出晶體過濾得目標化合物丙硫菌唑。其中,有機溶劑為甲醇、乙醇、異丙醇、乙腈、丙酮中一種或多種;其中,還原劑為TCEP、DTT、Zn、硼氫化鈉、氫化鋁鋰中的一種或幾種。
作為本發明所述丙硫菌唑的合成方法的一種優選方案,其中:所述有機溶劑,優選為甲醇。
作為本發明所述丙硫菌唑的合成方法的一種優選方案,其中:所述反應溫度,優選為30℃~50℃。
作為本發明所述丙硫菌唑的合成方法的一種優選方案,其中:所述還原劑,優選為金屬Zn。
本發明的有益效果:
(1)本發明合成工藝轉化率和選擇性高,合成原料便宜易得,降低了生產成本。
(2)反應條件溫和易控,操作簡便,產品提純容易,可以直接重結晶得到產物。
(3)各步中間體控制方法簡單、準確,產品收率較高,原子經濟性較好,避免繁瑣的後處理,具有很大的競爭優勢和工業生產利用價值。
(4)避免了使用強鹼等原料,三廢極低,符合綠色化學的理念。
附圖說明
為了更清楚地說明本發明實施例的技術方案,下面將對實施例描述中所需要使用的附圖作簡單地介紹,顯而易見地,下面描述中的附圖僅僅是本發明的一些實施例,對於本領域普通技術人員來講,在不付出創造性勞動性的前提下,還可以根據這些附圖獲得其它的附圖。其中:
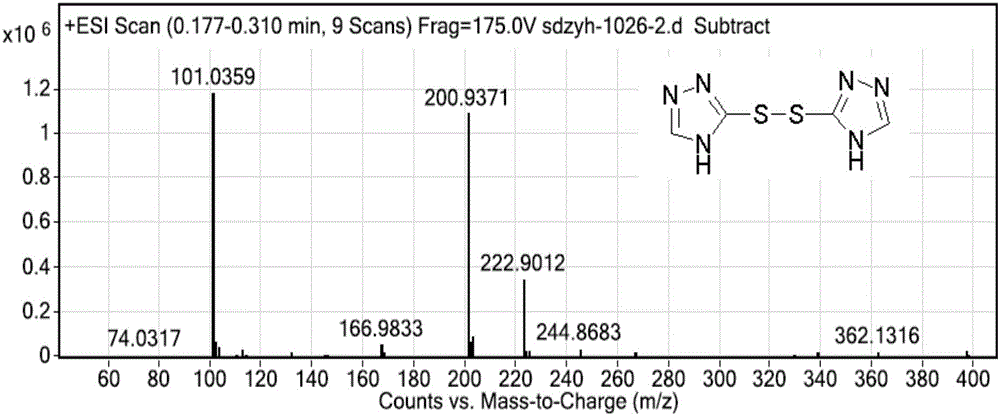
圖1為化合物Ⅳ的MS圖譜;
圖2為化合物Ⅴ的MS圖譜;
圖3為化合物Ⅵ的MS圖譜;
圖4為化合物Ⅶ的MS圖譜;
圖5為化合物Ⅶ的1H-NMR圖譜,其中,1H NMR(400MHz,Chloroform-d)δ11.92(s,1H),7.85(s,1H),7.59–7.50(m,1H),7.40–7.33(m,1H),7.21(tt,J=7.3,5.3Hz,2H),4.79(d,J=14.6Hz,1H),4.50(d,J=14.6Hz,1H),4.27(s,1H),3.62(d,J=14.0Hz,1H),3.17(d,J=14.0Hz,1H),1.02–0.85(m,2H),0.85–0.73(m,2H)。
具體實施方式
為使本發明的上述目的、特徵和優點能夠更加明顯易懂,下面結合具體實施例對本發明的具體實施方式做詳細的說明。
在下面的描述中闡述了很多具體細節以便於充分理解本發明,但是本發明還可以採用其他不同於在此描述的其它方式來實施,本領域技術人員可以在不違背本發明內涵的情況下做類似推廣,因此本發明不受下面公開的具體實施例的限制。
其次,此處所稱的「一個實施例」或「實施例」是指可包含於本發明至少一個實現方式中的特定特徵、結構或特性。在本說明書中不同地方出現的「在一個實施例中」並非均指同一個實施例,也不是單獨的或選擇性的與其他實施例互相排斥的實施例。
為了描述本發明的方便,使用各種試劑的常規和非常規縮寫。這些縮寫是本領域技術人員熟悉的,但是為了清楚在下面列出:
THF:四氫呋喃
DMF:N,N-二甲基甲醯胺
DMSO:二甲基亞碸
DCM:二氯甲烷
TCEP:三(2-羧乙基)膦
DTT:二硫蘇糖醇
實施例1:
2-(1-氯環丙基)-3-氯-1-(2-氯苯基)-2-丙醇的合成
在有恆壓滴定漏鬥和冷凝管的250mL圓底三口燒瓶裝置下,進行無水無氧操作氮氣保護條件下,將10g(0.062mol)2-氯氯苄與50mLTHF溶液混合置於恆壓滴液漏鬥中,在三口燒瓶中加入1.8g(0.074mol)鎂屑和10ml的THF溶液和少量的碘,滴入2ml 2-氯氯苄的THF溶液,微熱引發,將裝置放入冰浴中緩慢滴加(滴/3s)直至滴完,滴完後再反應1h,將上述格氏反應物慢慢滴入9.0g(0.058mol)1-氯-1-氯乙醯基環丙烷的THF(30mL)溶液緩慢滴加(滴/3s)直至滴完,滴完繼續反應1h,裝置一直放在冰浴中。最後緩慢將反應液緩慢加入到飽和的NH4Cl冰水溶液中淬滅反應,攪拌1h,用分液漏鬥分出有機相,加入無水硫酸鈉乾燥後,蒸除THF得到,得14.45g化合物(Ⅱ)淺黃色油狀液體,收率為89%。
實施例2:
丙硫菌唑的合成
化合物(Ⅳ)的合成
將5g(0.072mol)1,2,4-三氮唑溶於15ml的DMF中,加入6.9g(0.216mol)升華硫,加熱到140℃,反應3.5h後,冷卻至室溫,過濾,濾液用飽和氯化鈉洗滌,乙酸乙酯萃取,分出有機相,用無水硫酸鈉乾燥,蒸除乙酸乙酯得固體化合物(Ⅳ)6.95g,收率為95%。
化合物(Ⅴ)的合成
將3.03g(30mmol)化合物(Ⅳ)溶於30mLDCM中,加入2.37g(30mmol)重蒸吡啶。冰浴攪拌條件下,滴加2.64g(15mmol)苯磺醯氯,1h左右滴加完畢。移去冰水浴,室溫下攪拌5h。蒸去DCM,剩餘物加入15mL水和10mL乙酸乙酯,反應1h,過濾,並用適量的水和乙酸乙酯洗滌。將過濾得到的固體產物在真空乾燥箱裡抽乾,得到固體化合物(Ⅴ)2.88g,收率為96%。
化合物(Ⅵ)的合成
將4g(20mmol)化合物(Ⅴ)和20ml DMF,5.52g(40mmol)碳酸鉀攪拌混合,加熱至50℃。滴加化合物(Ⅱ)11.74g(42mmol),2h滴加完畢,繼續反應h,停止反應,冷卻至室溫。抽濾,濾液用飽和食鹽水洗滌,乙酸乙酯萃取,有機相用飽和食鹽水洗滌三次,分出有機相,無水硫酸鈉乾燥,旋蒸的產物。得到化合物(Ⅵ)11.4g,收率為83%。
目標化合物(Ⅶ)丙硫菌唑的合成
取5g(0.0072mol)化合物(Ⅵ)溶於30ml的甲醇中,加入0.97g(0.015mol)金屬Zn,30℃攪拌反應3h,停止反應,直接重結晶,析出晶體過濾得目標化合物(Ⅶ)丙硫菌唑4.3g,收率為86%。
本發明在上述實施例方式中,重新設計合成路線來降低生產成本,提高每一步的轉化率,其以1,2,4-三氮唑為原料經硫氧化得到巰基-1,2,4-三氮唑,再經過氧化形成二硫鍵,再與由1-氯-1-氯乙醯基環丙烷與2-氯氯苄經格式反應製得的2-(1-氯環丙烷)-3-氯-1-(2-氯苯基)-2-丙醇發生取代反應,再經還原就得到目標產物丙硫菌唑。其原理通過化學式表示為:
對比實施例1:
丙硫菌唑的合成
化合物(Ⅳ)的合成
將5g(0.072mol)1,2,4-三氮唑溶於15ml的DMSO中,加入11.52g(0.36mol)升華硫,加熱到180℃,反應5h後,冷卻至室溫,過濾,濾液用飽和氯化鈉洗滌,乙酸乙酯萃取,分出有機相,用無水硫酸鈉乾燥,蒸除乙酸乙酯得固體化合物(Ⅳ)5.89g,收率為81%。
化合物(Ⅴ)的合成
將3.03g(30mmol)化合物(Ⅳ)溶於30mLDCM中,加入3.16g(40mmol)重蒸吡啶。6℃攪拌條件下,滴加3.53g(20mmol)苯磺醯氯,1h左右滴加完畢。室溫下攪拌4h。蒸去DCM,剩餘物加入20mL水和20mL乙酸乙酯,反應1h,過濾,並用適量的水和乙酸乙酯洗滌。將過濾得到的固體產物在真空乾燥箱裡抽乾,得到固體化合物(Ⅴ)2.49g,收率為83%。
化合物(Ⅵ)的合成
將4g(20mmol)化合物(Ⅴ)和40ml乙腈,8.,28g(60mmol)碳酸鉀攪拌混合,加熱至80℃。滴加化合物(Ⅱ)21.24g(80mmol),2h滴加完畢,繼續反應4h,停止反應,冷卻至室溫。抽濾,濾液用飽和食鹽水洗滌,乙酸乙酯萃取,有機相用飽和食鹽水洗滌三次,分出有機相,無水硫酸鈉乾燥,旋蒸的產物。得到化合物(Ⅵ)10.7g,收率為78%。
目標化合物(Ⅶ)丙硫菌唑的合成
取5g(0.0072mol)化合物(Ⅵ)溶於30ml的乙醇中,加入1.98g(0.0079mol)TCEP,50℃攪拌反應3h,停止反應,直接重結晶,析出晶體過濾得目標化合物(Ⅶ)丙硫菌唑4.1g,收率為82%。
對比實施例2:
丙硫菌唑的合成
化合物(Ⅳ)的合成
將5g(0.072mol)1,2,4-三氮唑溶於20ml的甲苯中,加入9.22g(0.288mol)升華硫,加熱到100℃,反應10h後,冷卻至室溫,過濾,濾液用飽和氯化鈉洗滌,乙酸乙酯萃取,分出有機相,用無水硫酸鈉乾燥,蒸除乙酸乙酯得固體化合物(Ⅳ)4.07g,收率為56%。
化合物(Ⅴ)的合成
將3.03g(30mmol)化合物(Ⅳ)溶於30mLDCM中,加入3.16g(40mmol)重蒸吡啶。-2℃攪拌條件下,滴加3.53g(20mmol)苯磺醯氯,1h左右滴加完畢。室溫下攪拌5h。蒸去DCM,剩餘物加入20mL水和20mL乙酸乙酯,反應1h,過濾,並用適量的水和乙酸乙酯洗滌。將過濾得到的固體產物在真空乾燥箱裡抽乾,得到固體化合物(Ⅴ)2.1g,收率為70%。
化合物(Ⅵ)的合成
將4g(20mmol)化合物(Ⅴ)和40ml丙酮,11.04g(80mmol)碳酸鉀攪拌混合,加熱至30℃。滴加化合物(Ⅱ)21.24g(60mmol),2h滴加完畢,繼續反應4h,停止反應,冷卻至室溫。抽濾,濾液用飽和食鹽水洗滌,乙酸乙酯萃取,有機相用飽和食鹽水洗滌三次,分出有機相,無水硫酸鈉乾燥,旋蒸的產物。得到化合物(Ⅵ)7.0g,收率為51%。
目標化合物(Ⅶ)丙硫菌唑的合成
取5g(0.0072mol)化合物(Ⅵ)溶於30ml的丙酮中,加入0.55g(0.0144mol)硼氫化鈉,20℃攪拌反應5h,停止反應,直接重結晶,析出晶體過濾得目標化合物(Ⅶ)丙硫菌唑2.72g,收率為55%。
實施例3
化合物(Ⅵ)的合成
將6g(30mmol)化合物(Ⅴ)和30ml DMF,12.42g(90mmol)碳酸鉀攪拌混合,加熱至60℃。滴加化合物(Ⅱ)17.61g(63mmol),2h滴加完畢,繼續反應,停止反應,冷卻至室溫。抽濾,濾液用飽和食鹽水洗滌,乙酸乙酯萃取,有機相用飽和食鹽水洗滌三次,分出有機相,無水硫酸鈉乾燥,旋蒸的產物。得到化合物(Ⅵ)17.31g,收率為84%。
目標化合物(Ⅶ)丙硫菌唑的合成
取6.25g(0.009mol)化合物(Ⅵ)溶於30ml的甲醇中,加入1.21g(0.019mol)金屬Zn,40℃攪拌反應3h,停止反應,直接重結晶,析出晶體過濾得目標化合物(Ⅶ)丙硫菌唑5.5g,收率為88%。
實施例4
化合物(Ⅵ)的合成
將6g(30mmol)化合物(Ⅴ)和30ml DMF,12.42g(90mmol)碳酸鉀攪拌混合,加熱至80℃。滴加化合物(Ⅱ)17.61g(63mmol),2h滴加完畢,繼續反應,停止反應,冷卻至室溫。抽濾,濾液用飽和食鹽水洗滌,乙酸乙酯萃取,有機相用飽和食鹽水洗滌三次,分出有機相,無水硫酸鈉乾燥,旋蒸的產物。得到化合物(Ⅵ)16.49g,收率為80%。
目標化合物(Ⅶ)丙硫菌唑的合成
取6.25g(0.009mol)化合物(Ⅵ)溶於30ml的甲醇中,加入0.97g(0.015mol)金屬硼氫化鈉,50℃攪拌反應3h,停止反應,直接重結晶,析出晶體過濾得目標化合物(Ⅶ)丙硫菌唑5.06g,收率為81%。
實施例5
化合物(Ⅵ)的合成
將6g(30mmol)化合物(Ⅴ)和30ml DMF,12.42g(90mmol)碳酸鉀攪拌混合,加熱至100℃。滴加化合物(Ⅱ)17.61g(63mmol),2h滴加完畢,繼續反應,停止反應,冷卻至室溫。抽濾,濾液用飽和食鹽水洗滌,乙酸乙酯萃取,有機相用飽和食鹽水洗滌三次,分出有機相,無水硫酸鈉乾燥,旋蒸的產物。得到化合物(Ⅵ)11.54g,收率為56%。
目標化合物(Ⅶ)丙硫菌唑的合成
取6.25g(0.009mol)化合物(Ⅵ)溶於30ml的甲醇中,加入0.97g(0.015mol)金屬TECP,20℃攪拌反應3h,停止反應,直接重結晶,析出晶體過濾得目標化合物(Ⅶ)丙硫菌唑3.44g,收率為55%。
實施例3~5反應原理為:
在上述實施例1~5中,產生了合成目標化合物的關鍵中間體化合物,其化學結構式為:
基於上述實施例所提供的數據,可知本發明提供的丙硫菌唑的合成方法,能夠得到較為優異的收率。
在具體的實施例中,實施例1和2,均採用本發明提供的關於有機溶劑選擇、溫度控制範圍及還原劑選擇的優選方案,故而在每一階段目標化合物的收率均較為優異。相比之下,對比實施例1,基本遵循優選方案,而在合成路線中,個別目標化合物的合成中未採用本發明提供的溫度或還原劑的優選方案,相應得率均不理想。對比實施例2中,每一步目標化和物的合成,均未採用本發明關於反應時間、反應溫度、有機溶劑和還原劑選擇的優選範圍。
其中具體機理如下:
從反應溫度和反應時間角度看,過高的反應溫度及過長的反應時間,會出現反應平衡目標產物佔主要比例時,反應仍在繼續,平衡進一步推進,使得副產物增多;過低的反應溫度及過短的反應時間,會使得反應無法充分進行,並不能呈現較好的化學平衡,繼而獲得較佳的目標產物比收率。本發明通過對合成路線的設計,及每一步目標產物合成的反應溫度和反應時間的匹配設計,給出優選的控制範圍。
應說明的是,以上實施例僅用以說明本發明的技術方案而非限制,儘管參照較佳實施例對本發明進行了詳細說明,本領域的普通技術人員應當理解,可以對本發明的技術方案進行修改或者等同替換,而不脫離本發明技術方案的精神和範圍,其均應涵蓋在本發明的權利要求範圍當中。