一類苯並噻唑衍生物及其製備方法與流程
2023-05-10 18:02:56 3
本發明屬於新材料技術領域,具體涉及一類苯並噻唑衍生物及其製備方法。
(二)
背景技術:
有機螢光材料被廣泛應用於光學電子器件、DNA診斷、光化學傳感器、染料、螢光增白劑、螢光塗料、雷射染料、有機電致發光器件(OELD)等領域。與傳統的無機材料相比,有機非線性光學材料具有優異的光學性能、原料獲取廣泛、製備低成本、良好的可加工性等無可比擬的優勢,引起了很多人的關注,具有很大的潛在實用價值。
由於噻唑環S,N易配位的特性,噻唑環的衍生可用來製備離子探針、DNA探針,也可以用於分離元素rhenium(III),節省資源。苯並噻唑中硫原子易極化,使得分子軌道中電子易轉移,所以在光化學上作用出色。
苯並噻唑類衍生物的合成硫原子引入是關鍵步驟,選擇合適的反應原料對於苯並噻唑類衍生物的設計與合成來說至關重要。以鄰氨基苯硫酚製備苯並噻唑類衍生物,但難以獲得取代多樣的鄰氨基苯硫酚,隨後又出現硫代醯胺或硫脲.由於鄰氨基苯硫酚不穩定,很容易氧化,而硫代醯胺或硫脲這類原料的合成步驟比較長、製備成本比較高。
(三)、
技術實現要素:
本發明的第一個目的是提供一類苯並噻唑衍生物,是一類光學性能優異、穩定性好的有機螢光材料。本發明以苯並噻唑作為共軛單元,對其結構進行修飾得到了一類可在250-400nm範圍內具特定吸收的苯並噻唑衍生物。Jin報導了用2-(2-羥基-苯基)苯並噻唑衍生物檢測Zn2+,其stokes位移只有80nm,而本文所合成結構其stokes位移高達188nm。本發明採用的技術方案是:
本發明提供一類式(Ⅰ)所示苯並噻唑衍生物:
式(I)中:R1為氫、烷基或烷氧基;R2為氫或滷素原子。
作為優選,所述化合物為下列之一:
本發明的第二個目的是提供一種製備苯並噻唑衍生物(式Ⅰ所示化合物)的方法,所述製備方法包括如下步驟:
(1)將硫化試劑在溶劑A中回流1~2h後,將其加入到裝有溶劑A和化合物(II)的反應容器中,回流攪拌2~4h,過濾得到黃色固體(III),反應式如下:
(2)將化合物(III)與溶劑B和還原劑在100℃~120℃反應5~7h,等還原劑消耗完畢加入肉桂醛衍生物反應4h,然後用乙酸乙酯萃取,用體積比為15:1石油醚和乙酸乙酯過柱得到式(IV)所示的2-苯乙烯基苯並噻唑衍生物,反應式如下:
其中R2為氫或滷素原子;
(3)取化合物(IV)加入到反應容器中,加入鹼和化合物(IV)質量0.1倍的催化劑,然後再加入溶劑A,氬氣氛圍中回流24h後用體積比為10:1的石油醚和乙酸乙酯=過柱分離得到產物(V);所述鹼為碳酸鈉或碳酸鉀;催化劑為四(三苯基膦)鈀或二(三苯基膦)二氯化鈀;反應式如下:
其中R1為氫、烷基或烷氧基;R2為氫或滷素原子。
作為優選,步驟(1)中所述的硫化試劑選自硫、硫化鈉、硫化鉀和二氧化硫中的兩種;所述的溶劑A為甲醇、乙醇或丙醇,所述兩種硫化試劑與化合物(II)的物質的量比為1:1:2~4,所述硫化試劑在溶劑A的質量比為1:2.52:95.8~100,所述溶劑A和化合物(II)的質量比為8.8~9:1。
作為優選,步驟(2)中所述溶劑B為醋酸、鹽酸或硫酸;所述還原劑為鋅粉或錫粉,化合物(III)、還原劑和肉桂醛衍生物的摩爾比為1:89.6:1~2,所述化合物(III)與溶劑B的質量比為1:525~530。
作為優選,步驟(3)中所述溶劑A為乙醇、甲醇或醇;所述化合物(IV)、鹼和催化劑的物質的量比為1:8~10:0.1,所述溶劑A與化合物化合物(IV)的質量比為59~62:1。
本發明的第三個目的是提供一種苯並噻唑衍生物在螢光材料中的應用。本發明的目的是在苯並噻唑上引入溴原子,合成新型的苯並噻唑衍生物並應用到光學材料中。
本發明的有益效果:
本發明以苯並噻唑作為共軛單元,對其結構進行修飾,得到了一類在可250-400nm範圍內具特定吸收的苯並噻唑衍生物,所得苯並噻唑衍生物光學性能優異、穩定性好,能應用於螢光材料領域。
(四)附圖說明
圖1是為實施例1-4中製備的苯並噻唑衍生物在二氯甲烷中的紫外-可見吸收光譜圖,其中:橫坐標表示的是波長,單位為nm,縱坐標表示的是吸收強度,單位為1。
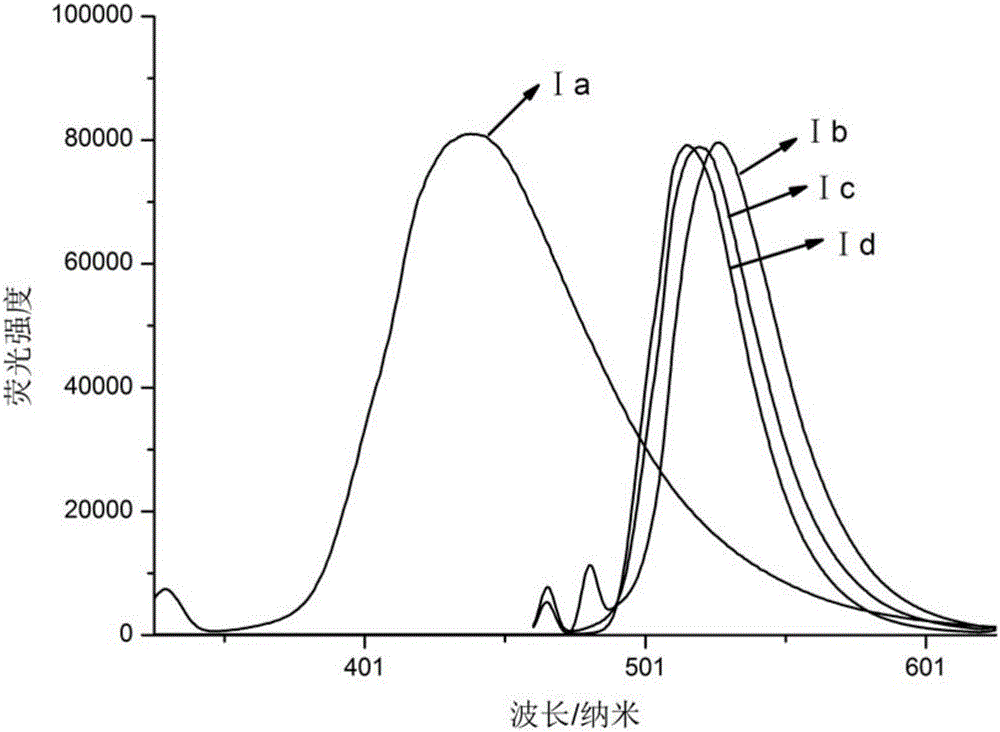
圖2是為實施例1-4中製備的苯並噻唑衍生物在二氯甲烷中的螢光光譜圖,其中:橫坐標表示的是波長,單位為nm,縱坐標表示的是強度,單位為1。
(五)具體實施方式
下面結合具體實施例對本發明進行進一步描述,但本發明的保護範圍並不僅限於此:
實施例1
(1)步驟一:
將4mmol的Na2S和4mmol的S在15ml甲醇溶液中回流1h後,用恆壓滴液漏鬥加入到裝有25ml甲醇和8mmol物質(II)的250ml圓底燒瓶中,回流攪拌4h,過濾得到黃色固體(III)(93mg),收率50%。
(2)步驟二:
將40mg的物質(III)加入到100ml圓底燒瓶中,加入20ml醋酸和1g鋅粉後110℃反應6h後,等鋅粉消耗完畢後加入11.33mg的肉桂醛衍生物反應4h,然後用乙酸乙酯萃取,用體積比為石油醚:乙酸乙酯=15:1過柱得到物質(IV)(2-苯乙烯基苯並噻唑衍生物),收率為50%。
(3)步驟三:
取物質(IV)1mmol加入到50ml三口燒瓶中,加入8mmol的K2CO3和10%當量的Pd(PPh3)4,加入乙醇25ml,氬氣氛圍中回流24h後用體積比為石油醚:乙酸乙酯=10:1過柱分離得到淡黃色固體產物(Va)2‐(4‐氟苯乙烯基)‐5‐((4‐甲氧基苯基)乙炔基)苯並噻唑,產率為60%。
m.p.183~185℃;1H NMR(500MHz,Chloroform-d)δ8.01(d,J=1.5Hz,1H),7.73(d,J=8.2Hz,1H),7.59–7.35(m,5H),7.35–7.15(m,1H),7.03(t,J=8.5Hz,2H),6.91–6.67(m,2H),3.76(s,3H).13C NMR(126MHz,CDCl3)δ167.56,159.55,153.51,136.70,133.84,132.90,131.31,131.28,129.08,129.01,128.33,125.23,121.65,121.34,121.25,115.96,115.78,114.88,113.87,89.64,87.52,77.28,77.22,77.03,76.77,55.11.HR-ESI/MS(m/z)calcd for C24 H17 F N O S[M+H]+=386.1009 found386.1022.
實施例2
(1)步驟一:
將4mmol的Na2S和4mmol的S在15ml甲醇溶液中回流1h後,用恆壓滴液漏鬥加入到裝有25ml甲醇和8mmol物質(II)的250ml圓底燒瓶中,回流攪拌4h,過濾得到黃色固體(III)(93mg),收率50%。
(2)步驟二:
將40mg的物質(III)加入到100ml圓底燒瓶中,加入20ml醋酸和1g鋅粉後110℃反應6h後,等鋅粉消耗完畢後加入11.33mg的肉桂醛衍生物反應4h,然後用乙酸乙酯萃取,用體積比為石油醚:乙酸乙酯=15:1過柱得到物質(IV)(2-苯乙烯基苯並噻唑衍生物),收率為50%。
(3)步驟三:
取物質(IV)1mmol加入到50ml三口燒瓶中,加入8mmol的K2CO3和10%當量的Pd(PPh3)4,加入乙醇25ml,氬氣氛圍中回流24h後用體積比為石油醚:乙酸乙酯=10:1過柱分離得到淡黃色固體產物(Vb)5-((4-甲氧基苯基)乙炔基)-2-苯乙烯基苯並噻唑,產率為60%。
m.p.170~172℃;1H NMR(500MHz,Chloroform-d)δ8.13(d,J=1.4Hz,1H),7.81(d,J=8.3Hz,1H),7.63–7.57(m,2H),7.57–7.50(m,4H),7.47–7.38(m,4H),7.28(s,0H),6.92(d,J=8.7Hz,2H),3.85(s,3H).13C NMR(126MHz,CDCl3)δ167.88,159.74,153.90,138.15,135.30,134.14,133.14,133.12,129.57,128.98,128.59,128.54,127.47,125.63,121.96,121.85,121.36,115.22,114.06,89.77,87.80,77.28,77.23,77.03,76.78,55.32.HR-ESI/MS(m/z)calcd for C24 H18 N O S[M+H]+=352.1154,found368.1094.
實施例3
(1)步驟一:
將4mmol的Na2S和4mmol的S在15ml甲醇溶液中回流1h後,用恆壓滴液漏鬥加入到裝有25ml甲醇和8mmol物質(II)的250ml圓底燒瓶中,回流攪拌4h,過濾得到黃色固體(III)(93mg),收率50%。
(2)步驟二:
將40mg的物質(III)加入到100ml圓底燒瓶中,加入20ml醋酸和1g鋅粉後110℃反應6h後,等鋅粉消耗完畢後加入11.33mg的肉桂醛衍生物反應4h,然後用乙酸乙酯萃取,用體積比為石油醚:乙酸乙酯=15:1過柱得到物質(IV)(2-苯乙烯基苯並噻唑衍生物),收率為50%。
(3)步驟三:
取物質(IV)1mmol加入到50ml三口燒瓶中,加入8mmol的K2CO3和10%當量的Pd(PPh3)4,加入乙醇25ml,氬氣氛圍中回流24h後用體積比為石油醚:乙酸乙酯=10:1過柱分離得到淡黃色固體產物(Vc)2-苯乙烯基-5-(對甲苯基乙基)苯並噻唑,產率為60%。
m.p.166~168℃;1H NMR(500MHz,Chloroform-d)δ8.13(dd,J=3.9,1.4Hz,1H),7.80(dd,J=16.4,8.3Hz,1H),7.62–7.58(m,2H),7.57–7.51(m,1H),7.51–7.45(m,2H),7.45–7.36(m,3H),7.36–7.22(m,1H),7.18(dd,J=8.2,2.8Hz,2H),2.39(d,J=2.0Hz,4H),13C NMR(126MHz,CDCl3)δ167.88,153.87,138.54,138.16,135.26,134.28,131.54,131.52,129.56,129.13,128.96,128.85,128.57,128.43,128.41,127.99,127.45,126.45,125.71,125.50,121.92,121.68,121.39,121.37,120.00,89.92,88.41,77.28,77.23,77.03,76.77,21.51.HR-ESI/MS(m/z)calcd for C24 H18 N S[M+H]+=352.1154,found352.1146.
實施例4
(1)步驟一:
將4mmol的Na2S和4mmol的S在15ml甲醇溶液中回流1h後,用恆壓滴液漏鬥加入到裝有25ml甲醇和8mmol物質(II)的250ml圓底燒瓶中,回流攪拌4h,過濾得到黃色固體(III)(93mg),收率50%。
(2)步驟二:
將40mg的物質(III)加入到100ml圓底燒瓶中,加入20ml醋酸和1g鋅粉後110℃反應6h後,等鋅粉消耗完畢後加入12.88mg的肉桂醛衍生物反應4h,然後用乙酸乙酯萃取,用體積比為石油醚:乙酸乙酯=15:1過柱得到物質(IV)(2-苯乙烯基苯並噻唑衍生物),收率為50%。
(3)步驟三:
取物質(IV)1mmol加入到50ml三口燒瓶中,加入8mmol的K2CO3和10%當量的Pd(PPh3)4,加入乙醇25ml,氬氣氛圍中回流24h後用體積比為石油醚:乙酸乙酯=10:1過柱分離得到淡黃色固體產物(Vd)5-(苯基乙炔基)-2-苯乙烯基苯並噻唑,產率為57.4%。
m.p.160~163℃;1H NMR(500MHz,Chloroform-d)δ8.16(d,J=1.5Hz,1H),7.84(d,J=8.2Hz,1H),7.62(d,J=1.6Hz,1H),7.61(d,J=2.1Hz,2H),7.59(d,J=1.7Hz,1H),7.57(s,0H),7.56(d,J=1.5Hz,1H),7.54(d,J=1.6Hz,1H),7.46–7.43(m,2H),7.42(d,J=4.2Hz,1H),7.41(d,J=1.2Hz,0H),7.40–7.37(m,3H),7.28(s,1H).13C NMR(126MHz,CDCl3)δ167.96,153.90,138.24,135.28,134.50,131.68,129.60,128.99,128.60,128.39,127.48,125.85,123.13,121.93,121.49,121.43,89.73,89.08,77.28,77.02,76.77.HR-ESI/MS(m/z)calcd for C23 H16 N S[M+H]+=338.0988,found338.0998.
實施例5
(1)步驟一:
將4mmol的K2S和4mmol的SO2在15ml乙醇溶液中回流2h後,用恆壓滴液漏鬥加入到裝有25ml乙醇和16mmol物質(II)的250ml圓底燒瓶中,回流攪
拌2h,過濾得到黃色固體(III),收率16.8%。
(2)步驟二:
將40mg的物質(III)加入到100ml圓底燒瓶中,加入20ml鹽酸和1.8g錫粉,然後在130℃反應8h,等錫粉消耗完畢後加入22.66mg的肉桂醛衍生物反應4h,然後用乙酸乙酯萃取,用體積比為石油醚:乙酸乙酯=15:1過柱得到物質(IV)(2-苯乙烯基苯並噻唑衍生物),收率為15%。
(3)步驟三:
取物質(IV)1mmol加入到50ml三口燒瓶中,加入10mmol的Na2CO3和10%當量的[(C6H5)3P]2PdCl,加入丙醇25ml,氬氣氛圍中回流24h後用體積比為石油醚:乙酸乙酯=10:1過柱分離得到淡黃色固體產物(Ve)5-(苯基乙炔基)-2-苯乙烯基苯並噻唑,產率為11.4%。
實施例6
(1)步驟一:
將4mmol的K2S和4mmol的SO2在15ml乙醇溶液中回流2h後,用恆壓滴液漏鬥加入到裝有25ml乙醇和16mmol物質(II)的250ml圓底燒瓶中,回流攪拌2h,過濾得到黃色固體(III),收率16.8%。
(2)步驟二:
將40mg的物質(III)加入到100ml圓底燒瓶中,加入20ml鹽酸和1.8g錫粉,然後在130℃反應8h,等錫粉消耗完畢後加入22.66mg的肉桂醛衍生物反應4h,然後用乙酸乙酯萃取,用體積比為石油醚:乙酸乙酯=15:1過柱得到物質(IV)(2-苯乙烯基苯並噻唑衍生物),收率為15%。
(3)步驟三:
取物質(IV)1mmol加入到50ml三口燒瓶中,加入10mmol的Na2CO3和10%當量的[(C6H5)3P]2PdCl,加入丙醇25ml,氬氣氛圍中回流24h後用體積比為石油醚:乙酸乙酯=10:1過柱分離得到產物(Vf)苯並噻唑衍生物,產率為14.4%。
實施例7
(1)步驟一:
將4mmol的K2S和4mmol的SO2在15ml乙醇溶液中回流2h後,用恆壓滴液漏鬥加入到裝有25ml乙醇和16mmol物質(II)的250ml圓底燒瓶中,回流攪拌2h,過濾得到黃色固體(III),收率16.8%。
(2)步驟二:
將40mg的物質(III)加入到100ml圓底燒瓶中,加入20ml鹽酸和1.8g錫粉,然後在130℃反應8h,等錫粉消耗完畢後加入22.66mg的肉桂醛衍生物反應4h,然後用乙酸乙酯萃取,用體積比為石油醚:乙酸乙酯=15:1過柱得到物質(IV)(2-苯乙烯基苯並噻唑衍生物),收率為15%。
(3)步驟三:
取物質(IV)1mmol加入到50ml三口燒瓶中,加入10mmol的Na2CO3和10%當量的[(C6H5)3P]2PdCl,加入丙醇25ml,氬氣氛圍中回流24h後用體積比為石油醚:乙酸乙酯=10:1過柱分離得到產物(Vg)苯並噻唑衍生物,產率為12.6%。
實施例8
(1)步驟一:
將4mmol的K2S和SO2在15ml乙醇溶液中回流2h後,用恆壓滴液漏鬥加入到裝有25ml乙醇和16mmol物質(II)的250ml圓底燒瓶中,回流攪拌2h,過濾得到黃色固體(III),收率16.8%。
(2)步驟二:
將40mg的物質(III)加入到100ml圓底燒瓶中,加入20ml鹽酸和1.8g錫粉,然後在130℃反應8h,等錫粉消耗完畢後加入36.22mg的肉桂醛衍生物反應4h,然後用乙酸乙酯萃取,用體積比為石油醚:乙酸乙酯=15:1過柱得到物質(VI)(2-苯乙烯基苯並噻唑衍生物),收率為13.4%。
(3)步驟三:
取物質(VI)1mmol加入到50ml三口燒瓶中,加入10mmol的Na2CO3和10%當量的[(C6H5)3P]2PdCl,加入丙醇25ml,氬氣氛圍中回流24h後用體積比為石油醚:乙酸乙酯=10:1過柱分離得到產物(Vh)苯並噻唑衍生物,產率為12.4%。
實施例9
苯並噻唑衍生物的性能檢測
紫外-可見吸收光譜見圖1:將化合物配成濃度為C=4×10-6mol/L的溶液,溶劑為二氯甲烷,使用的儀器是Shimadzu UV-1800分光光度計。從圖1紫外吸收光譜中可以看出,4a-4d的紫外光譜均呈現兩個明顯的最大吸收峰,出現在290nm處的為π→π*電子躍遷產生的吸收峰;出現在310nm左右吸收峰則源於分子與CH2Cl2溶劑作用吸收。螢光關譜見圖2:將化合物配成濃度為C=4×10-6mol/L的溶液,溶劑為二氯甲烷,使用的儀器是Shimadzu RF-6000PC光譜儀。從圖2螢光發射圖中看出Ⅰa分子為甲氧基與氟的衍生取代,相比Ⅰb,Ⅰc,Ⅰd發射峰520nm,發射光譜Ⅰa藍移,最大發射峰在450左右。由於Ⅰa中F為吸電子基,電子云集中,導致共軛平面改變,發生藍移。
表1苯並噻唑衍生物紫外吸收與螢光
λmax表示最大吸收波長,Eex表示激發波長,Em表示發射波長,Stokes shift表示斯託克斯位移。
由於Ia中F為吸電子基,電子云集中,導致共軛平面改變,發生藍移,導致stokes位移小於Ib,Ic,Id。
以上僅列舉了本發明的優選實施方案,本發明的保護範圍並不限制於此,本領域技術人員在本發明權利要求範圍內所作的任何改變均落入本發明保護範圍內。