一種豬嵴病毒全長cDNA的構建方法與流程
2023-07-08 05:34:11 4
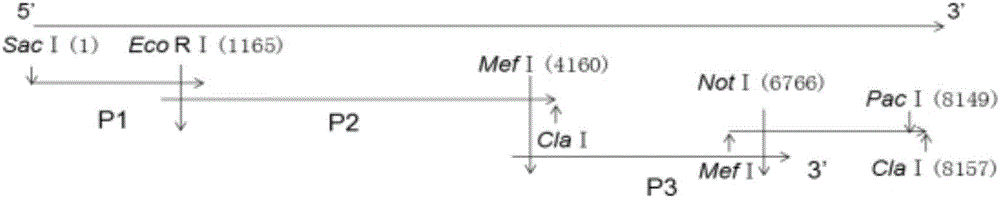
本發明涉及生物
技術領域:
,具體涉及一種豬嵴病毒全長cDNA的構建方法。
背景技術:
:近年來,反向遺傳操作技術在病毒學研究中取得了一定進展。構建感染性cDNA後通過體內或體外的方式獲得病毒RNA的方法,在RNA病毒的研究中將發揮著越來越重要的作用。對病毒基因進行修飾拯救出具有生物活性的病毒後將其與野生型病毒進行比較,能為病毒病的防制及疫苗研究提供思路。豬嵴病毒的研究進程因未找到合適的病毒分離及純化方法而受到嚴重阻礙。技術實現要素:鑑於現有技術的缺陷,本發明旨在於提供一種豬嵴病毒全長cDNA的構建方法。為獲的純化的病毒,本發明通過分析從流行病學調查中選出的在腹瀉仔豬中較流行的PKVCH441株基因組和pBlueScriptKS(Ⅱ)載體結構,對PKVCH441株基因組進行了分段RT-PCR擴增並克隆在pBlueScriptKS(Ⅱ)載體上,成功構建了包含有PKV全長的質粒,該研究為了解PKV在細胞中的複製機制,研製疫苗奠定了夯實的基礎。為了實現上述目的,本發明採用的技術方案如下:1、一種豬嵴病毒全長cDNA的構建方法,其特徵在於,所述方法包括如下步驟:S1利用Primerpremier5軟體對拼接後CH441株全基因組序列和pBlueScriptKS(Ⅱ)載體進行序列分析,選擇了載體和全基因組中都存在的單一酶切位點EcoRⅠ和NotⅠ,在全基因組中單一存在的MefⅠ及載體上不存在但通過引物在序列3』末端引入的PacⅠ酶切位點以及雖然全基因組序列上不止一個但可以通過調整順序插入的SacⅠ和ClaⅠ酶切位點;利用這些酶切位點將全基因組連接在同一載體上;S2片段P1與pBlueScriptKS(Ⅱ)載體連接:將已克隆到pMD19-TSimpleVector載體上測序正確的pMD19-T-P1質粒用SacⅠ和EcoRⅠ限制性內切酶雙酶切,反應體系為:10×NEBbuffer5μL;SacⅠ和EcoRⅠ限制性內切酶各2μL;pMD19-T-P1質粒10μL;去離子水31μL,混勻後37℃作用2h,回收後的片段與用相同的限制性內切酶酶切的pBlueScriptKS(Ⅱ)純化回收產物用T4DNA連接酶進行連接,10μL反應體系如下:2×T4DNA連接酶1μL,T4DNA連接酶buffer1μL,載體1μL,P1片段7μL,混勻並16℃連接過夜後進行轉化;次日挑取單菌落培養後,選出雙酶切鑑定呈陽性的質粒送測序;將測序結果與已獲得的PKVCH441株對應序列進行比對分析,測序正確的質粒命名為pBKS-P1;S3P2片段與pBKS-P1的連接:測序正確的pBKS-P1用EcoRⅠ和ClaⅠ限制性內切酶雙酶切處理。回收後的片段與用相同方法處理的pMD19-T-P2回收產物用T4DNA連接酶進行連接,連接過夜後進行轉化。挑選單菌落培養後提取質粒,選出雙酶切鑑定呈陽性的質粒送測序;測序正確的質粒命名為pBKS-P1-P2;S43』片段與pBKS-P1-P2的連接:測序正確的pBKS-P1-P2用MefⅠ和ClaⅠ限制性內切酶雙酶切處理。目的片段回收後與用相同方法處理的pMD19-T-3』回收產物用T4DNA連接酶連接過夜後進行轉化;挑選單菌落培養後提取質粒,選出雙酶切鑑定呈陽性的質粒送測序;將測序結果與已測得的PKVCH441株對應序列進行比對分析,測序正確者命名為pBKS-P1-P2-3』;S5P3片段與pBKS-P1-P2-3』的連接:測序正確的pBKS-P1-P2-3』用MefⅠ和NotⅠ限制性內切酶雙酶切處理。回收後的片段與用相同方法處理的pMD19-T-P3回收產物連接過夜後進行轉化;挑選單菌落培養適當時間後提取質粒,選出雙酶切鑑定呈陽性的質粒送測序;測序結果與已測得的PKVCH441株對應序列進行比對分析,測序正確的質粒命名為pBKS-P1-P2-3』-P3;S6將測序合適的pBKS-P1-P2-3』-P3質粒用PacⅠ限制性內切酶單酶切,酶切反應在50μL體系中進行:10×NEBbuffer5μL;PacⅠ5μL;pBKS-P1-P2-3』-P3質粒10μL;去離子水30μL,混勻後37℃作用2h,然後進行純化回收;S7回收的片段進行體外轉錄。所述純化回收的步驟為:(1)將PCR產物轉移到新的1.5mL離心管中,加入5倍體積的BufferCP;(2)將上述混合液混勻後轉移到試劑盒提供的吸附柱中,10000g離心1min,棄濾液;(3)向吸附柱中加入700μLDNAWashBuffer,10000g離心1min,棄濾液;(4)重複步驟(3)一次;(5)全速離心2min;(6)將吸附柱套入新的1.5mL離心管,向吸附柱中垂直加入30μL洗脫液,靜止2min;(7)全速離心2min,收集DNA。所述體外轉錄的步驟為:(1)將混合液輕輕混勻,37℃作用2h~4h;(2)向混合液中加入1μLTURBODNase,混勻後37℃孵育15min;轉錄後的混合液用RNeasyMinEluteCleanupKit純化回收,步驟如下:(1)用DEPC水調整混合液的體積到450μL。(2)在上述混合液中加入250μL無水乙醇,輕輕混勻。(3)將上述混合液轉入2mL的RNeasyMinElute管中,8000g離心15s後棄取液體。(4)將上述RNeasyMinElute收集管轉入新的2mL柱子中,加入500μLRPE後8000g離心15s後棄去液體。(5)在上述混合液中加入500μL80%無水乙醇,輕輕混勻,8000g離心2min後棄液體和收集管。(6)將RNeasyMinElute柱子轉移進入2mL離心管中,全速離心5min,棄收集管。(7)將RNeasyMinElute柱子轉移進入1.5mL離心管中,加入14μLDEPC水,全速離心1min後收集液體。(8)回收後的RNA於-80℃保存備用。本發明有益效果在於,本研究通過RNA病毒的經典研究方法-反向遺傳技術,成功構建了PKVCH441株全長質粒,並進行了體外轉錄,為其進一步深入研究PKV夯實了堅定的基礎。附圖說明圖1為本發明PKVCH441基因組酶切位點選擇示意圖;圖2為本發明豬嵴病毒全長質粒構建示意圖;圖3為本發明PKVCH441全基因組各片段的擴增;圖4為不同PKV全基因組核苷酸序列相似性分析;圖5為不同PKV全基因組進化樹分析;圖6為本發明的豬嵴病毒CH441全長質粒酶切鑑定圖;具體實施方式下面通過具體的實施方式進一步對本發明進行描述,需要說明的是,所涉及的實施方式/實施例只是便於描述本發明的技術方案,並不是對本發明的限制。關於病毒、載體與菌株豬嵴病毒株CH441和pBlueScriptKS(Ⅱ)載體由中國農業科學院蘭州獸醫研究所動物病毒分子生態學創新團隊保存;大腸桿菌DH5а受體菌和pMD19-TSimpleVector均購自寶生物工程(大連)有限公司。關於主要試劑和儀器主要試劑:MEGAscriptKit購自invitrogen公司;PlasmidMiniKitⅠ、GelExtractionKitⅠ及Cycle-PureKit均購自Omega公司;MiniBESTUniversalRNAExtractionKit試劑盒、OneStepPrimeScriptRT-PCRKit試劑盒、DL2000DNAMarker和DL5000DNAMarker均購自寶生物工程(大連)有限公司;T4DNA連接酶及限制性內切酶均購自美國紐英倫生物技術(北京)有限公司。主要儀器:移液槍購自Eppendorf公司;離心機購自Sigma公司;PCR儀購自德國Eppendorf公司;核酸電泳系統、UniversalHoodII凝膠成像系統均購自美國Bio-Rad公司;製冰機購自日本三洋公司;恆溫搖床購自上海智城公司;恆溫金屬浴購自天根生化技術(北京)有限公司。關於PKVCH441全基因組擴增引物設計由於PKVCH4415』端序列高度保守且實驗室已通過5』Race完成了其序列測定工作,在此基礎上,我們將CH441株豬嵴病毒全基因組分為4段進行全基因RT-PCR擴增。擴增引物設計如下:關於全基因組分段擴增及測序1、重組質粒的構建回收目的基因片段克隆到pMD19-TSimpleVector。混勻體系後在恆溫金屬浴上16℃連接過夜後轉化連接產物。次日從平板上挑取幾個單菌落,培養適當時間後提取質粒。酶切鑑定陽性後送華大基因公司測序。關於全基因組序列的拼接與分析測序鑑定正確後,將包含PKVCH441全序列的4個片段進行序列比對分析,分別選出4個片段的優勢序列,將選出的序列用DNAStar軟體進行拼接獲得CH441全基因組序列,將後者與登錄在GenBank中的PKV全基因組序列進行比對分析。關於限制性內切酶位點的選擇如圖1所示,利用Primerpremier5軟體對拼接後CH441株全基因組序列和pBlueScriptKS(Ⅱ)載體進行序列分析,選擇了載體和全基因組中都存在的單一酶切位點EcoRⅠ和NotⅠ,在全基因組中單一存在的MefⅠ及載體上不存在但通過引物在序列3』末端引入的PacⅠ酶切位點以及雖然全基因組序列上不止一個但可以通過調整順序插入的SacⅠ和ClaⅠ酶切位點。利用這些酶切位點將全基因組連接在同一載體上。關於CH441全基因組分段如圖2所示將已克隆到pMD19-TSimpleVector上的測序合適的質粒按合適的順序和酶切位點酶切並連接到pBlueScriptKS(Ⅱ)載體上,構建PKVCH441全長質粒。片段P1與pBlueScriptKS(Ⅱ)載體連接:將已克隆到pMD19-TSimpleVector載體上測序正確的pMD19-T-P1質粒用SacⅠ和EcoRⅠ限制性內切酶雙酶切,反應體系為:10×NEBbuffer5μL;SacⅠ和EcoRⅠ限制性內切酶各2μL;pMD19-T-P1質粒10μL;去離子水31μL,混勻後37℃作用2h,回收後的片段與用相同的限制性內切酶酶切的pBlueScriptKS(Ⅱ)純化回收產物用T4DNA連接酶進行連接,10μL反應體系如下:2×T4DNA連接酶1μL,T4DNA連接酶buffer1μL,載體1μL,P1片段7μL,混勻並16℃連接過夜後進行轉化。次日挑取單菌落培養後用PlasmidMiniKitI提取質粒,選出雙酶切鑑定呈陽性的質粒送測序。將測序結果與已獲得的PKVCH441株對應序列進行比對分析,測序正確的質粒命名為pBKS-P1。P2片段與pBKS-P1的連接:測序正確的pBKS-P1用EcoRⅠ和ClaⅠ限制性內切酶雙酶切處理。回收後的片段與用相同方法處理的pMD19-T-P2回收產物用T4DNA連接酶進行連接,連接過夜後進行轉化。挑選單菌落培養後提取質粒,選出雙酶切鑑定呈陽性的質粒送測序。測序正確的質粒命名為pBKS-P1-P2。3』片段與pBKS-P1-P2的連接:測序正確的pBKS-P1-P2用MefⅠ和ClaⅠ限制性內切酶雙酶切處理。目的片段回收後與用相同方法處理的pMD19-T-3』回收產物用T4DNA連接酶連接過夜後進行轉化。挑選單菌落培養後提取質粒,選出雙酶切鑑定呈陽性的質粒送測序。將測序結果與已測得的PKVCH441株對應序列進行比對分析,測序正確者命名為pBKS-P1-P2-3』。P3片段與pBKS-P1-P2-3』的連接:測序正確的pBKS-P1-P2-3』用MefⅠ和NotⅠ限制性內切酶雙酶切處理。回收後的片段與用相同方法處理的pMD19-T-P3回收產物連接過夜後進行轉化。挑選單菌落培養適當時間後提取質粒,選出雙酶切鑑定呈陽性的質粒送測序。測序結果與已測得的PKVCH441株對應序列進行比對分析,測序正確的質粒命名為pBKS-P1-P2-3』-P3。關於PKVCH441的體外轉錄將測序合適的pBKS-P1-P2-3』-P3質粒用PacⅠ限制性內切酶單酶切,酶切反應在50μL體系中進行:10×NEBbuffer5μL;PacⅠ5μL;pBKS-P1-P2-3』-P3質粒10μL;去離子水30μL,混勻後37℃作用2h,然後根據Cycle-PureKit說明書進行純化回收。純化回收步驟如下:(1)將PCR產物轉移到新的1.5mL離心管中,加入5倍體積的BufferCP;(2)將上述混合液混勻後轉移到試劑盒提供的吸附柱中,10000g離心1min,棄濾液;(3)向吸附柱中加入700μLDNAWashBuffer,10000g離心1min,棄濾液;(4)重複步驟(3)一次;(5)全速離心2min;(6)將吸附柱套入新的1.5mL離心管,向吸附柱中垂直加入30μL洗脫液,靜止2min;(7)全速離心2min,收集DNA。回收的片段按MEGAscriptKitprocedure說明體外轉錄,體系如下:AmountComponentto20μLNuclease-freeWater2μLATPsolution2μLCTPsolution2μLGTPsolution2μLUTPsolution2μL10×ReactionBuffer0.1-1μgLineartemplateDNA+2μLEnzymeMix步驟為:(1)將上述混合液輕輕混勻,37℃作用2h~4h;(2)向上述混合液中加入1μLTURBODNase,混勻後37℃孵育15min;轉錄後的混合液用RNeasyMinEluteCleanupKit純化回收,步驟如下:(1)用DEPC水調整混合液的體積到450μL。(2)在上述混合液中加入250μL無水乙醇,輕輕混勻。(3)將上述混合液轉入2mL的RNeasyMinElute管中,8000g離心15s後棄取液體。(4)將上述RNeasyMinElute收集管轉入新的2mL柱子中,加入500μLRPE後8000g離心15s後棄去液體。(5)在上述混合液中加入500μL80%無水乙醇,輕輕混勻,8000g離心2min後棄液體和收集管。(6)將RNeasyMinElute柱子轉移進入2mL離心管中,全速離心5min,棄收集管。(7)將RNeasyMinElute柱子轉移進入1.5mL離心管中,加入14μLDEPC水,全速離心1min後收集液體。(8)回收後的RNA於-80℃保存備用。關於全基因組各片段的擴增結果如圖3所示,包含CH441全長的4個片段擴增結果。P1、P2、P3及3』大小分別為1265bp、3099bp、2673bp和1429bp。通過1.0%瓊脂糖核酸凝膠分析各片段擴增結果與預期結果相符。關於CH441全基因組序列比對分析結果序列拼接結果顯示,PKVCH441株全基因組全長為8148nt,A+T百分含量為47.83%,G+C百分比為52.17%。基因組成如下表:如圖4、圖5所示,利用MegAlign軟體對NCBI中提交的部分PKV全基因組與PKVCH441株進行比對分析,結果顯示,核苷酸相似度在到87.9%~90.8%之間,並且2B區域缺失90nt,這可能與自然選擇和病原與宿主間的相互作用有關。關於構建全長cDNA雙酶切鑑定圖如圖6所示,pBKS-P1雙酶切片段分別為3000bp和1265bp;pBKS-P1-P2雙酶切片段分別為4162bp和3000bp;pBKS-P1-P2-3』雙酶切片段分別為7162bp和1429bp;pBKS-P1-P2-3』-P3雙酶切片段分別為10162bp和2699bp;與預期結果一致,說明全長質粒構建成功。線性化的全長質粒體外轉錄後純化的RNA濃度可達4000ng/μL反向遺傳操作技術即利用現代生物學理論與方法,通過對核苷酸水平的操作來研究生物表型變化的一種手段,它與經典的從表型改變到基因功能研究的思路相反。目前採用的根據病毒的核苷酸序列,運用反向遺傳操作技術來拯救RNA病毒的方法不僅可以克服體內RNA複製酶保真性低的缺點而且可為製備高免疫源性的低致病力毒株疫苗提供新的途徑,同時能為研究RNA病毒的致病機制提供了一種有效手段。RACE技術即cDNA末端快速擴增技術又稱錨定PCR,是由已知cDNA序列,擴增完整5』和3』末端的一種簡便、有效的方法,包括3』race和5』race兩種。同5』race相比,3』race更易成功且擴增片段長度更長。其基本原理為:利用mRNA的3』末端的多聚腺苷酸尾巴作為引物結合的位點,以通用接頭引物的Oligo(dT)為引物反轉錄成cDNA。然後以反轉錄得到的cDNA為模板,特異引物GSP1和含有部分接頭序列的通用引物UPM為上、下遊引物進行PCR擴增。5』race第一步同3』race不同之處為:反轉錄達到cDNA5』末端時自動加上3~5個胞嘧啶殘基,後者可以5』raceadapterprimer鹼基配對,轉換為含有SMART接頭序列的cDNA為模板,然後以該cDNA為模板,含有部分接頭序列的通用引物UPM和基因特異引物GSP2為上、下遊引物,進行PCR擴增。隨著PCR克隆技術的完善及RACE技術的不斷改進和發展,RACE技術在基因克隆及基因表達研究中的作用將越來越顯著。pBlueScriptKS(Ⅱ)是將T3、T7或SP6噬菌體啟動子序列,組入噬菌體載體的多克隆位點區兩側,構建而成的體外轉錄載體。在體外系統中,加入合適的噬菌體RNA聚合酶就可使插入該載體的外源基因得到轉錄。由於RNA聚合酶的活性具有高度的專一性,它們只能識別噬菌體自己啟動子的特定序列所以不會發生質粒基因的轉錄。其結構特點為:小分子量的共價、閉合、環狀的基因組DNA,T3和T7噬菌體啟動子分別位於多克隆位點的兩側,可定向啟動外源基因的轉錄活性。該載體的特點為:含有氨苄青黴素抗性基因,可作為轉化克隆的選擇標記;拷貝數高;存在多克隆位點,可克隆達10kb的外源基因;可用組織化學顯色反應法篩選噬菌體載體的重組子;含有質粒複製起點,即使在無輔助質粒存在的情況下,克隆的外源基因也可複製。因此,它不僅可以用於製備篩選cDNA文庫或者基因組文庫的放射性同位素標記的DNA分子探針;其體外轉錄合成的轉錄本還可以用來編碼具有外源基因正確讀碼結構的外源蛋白。本研究通過RNA病毒的經典研究方法-反向遺傳技術,成功構建了PKVCH441株全長質粒,並進行了體外轉錄,為其進一步深入研究PKV夯實了堅定的基礎。對於本領域的技術人員來說,可根據以上描述的技術方案以及構思,做出其它各種相應的改變以及變形,而所有的這些改變以及變形都應該屬於本發明權利要求的保護範圍之內。當前第1頁1 2 3