一種具有高生物活性的天然活性藥物中間體及其製備方法與流程
2023-07-15 21:24:01 2
本發明涉及一種具有高生物活性的天然活性藥物中間體及其製備方法,屬醫藥領域。
背景技術:
迄今為止,大量的天然存在的生物活性化合物已經在許多文獻中描述。特別是具有特別活性功能結構的生物活性中間體,例如類黃酮衍生的紫檀烷和稠合環類的含二氫吲哚的吡咯烷基二氫吲哚已經受到科研學者的重視。
紫檀烷,是第二大類天然異黃酮類化合物,是在具有6a,11a-二氫-6H-苯並呋喃並[3,2-c]色烯骨架的天然植物主要構架,其具有罕見的二氫苯並呋喃和苯並吡喃二環連接結構。其之所以受到科研工作者的廣泛關注主要是由於其具有一系列的生物表徵活性,例如COX-2抑制,LDL抗氧化,抗HIV等生物學特性。
吡咯烷基二氫吲哚, 是具有稠合二氫吲哚基團的典型的天然物中間體,主要存在於天然存在的具有次氯酸乙醯膽鹼酯酶抑制劑生物特性的(如毒扁豆鹼)天然植物中間體化合物中。在大多數情況下,吡咯烷基二氫吲哚的稠合二氫吲哚環體系可以通過吲哚或羥吲哚中間體以逐步連續合成法獲得,然後進一步製備為吡咯烷基二氫吲哚的目標產物。
以上兩種物質雖然最近已經報導了許多合成方法,但是其缺點在於起合成方法均為多步合成法,製備過程步驟多而繁瑣,且其總收率低,前者不足25%,後者不足65%,並且多依賴於稀有金屬催化劑作為反應中間催化劑。
技術實現要素:
基於上述情況,本發明的目的在於提供一種製備過程簡單,方便,所得產品安全無副作用的高生物活性的天然活性藥物中間體的製備方法。
一種具有高生物活性的天然活性藥物中間體,其特徵在於:所述天然活性藥物中間體為紫檀烷類化合物或吡咯烷基二氫吲哚類化合物,所述紫檀烷類化合物的結構式為:;所述吡咯烷基二氫吲哚類化合物的結構式為:。
優選的,所述具有高生物活性的天然活性藥物中間體的製備方法為:將原料在高價碘氧化劑(PIDA)和醇類溶劑的作用下,合成得到反應底物;再將反應底物和親核試劑溶解在二氯甲烷(DCM)中,然後加入六氟異丙醇(HFIP)和五氟苯甲酸(PFBA),反應完成後蒸發濃縮提純反應液,最終得到產品。
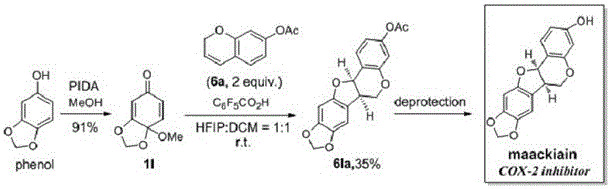
優選的,所述具有高生物活性的天然活性藥物中間體的製備方法,包括所述紫檀烷類化合物的製備:將原料苯酚類化合物在高價碘氧化劑(PIDA)和甲醇的作用下,合成得到反應底物醌單縮醛(QMA) 1l;再將1-3mmol醌單縮醛和親核試劑2H-苯並吡喃-7-基乙酸酯6a的混合物溶解在2-3ml的二氯甲烷(DCM)中,然後加入2-3ml六氟異丙醇(HFIP)和0.5-1.5mmol五氟苯甲酸(PFBA),在室溫下攪拌並用矽膠點板TLC監測測反應進程,待反應完成後再經過蒸發濃縮,然後通過快速矽膠色譜法(己烷/乙酸乙酯= 4/1)從反應濃縮液中直接進行產物的分離,最終得到產品紫檀烷6la。
進一步的,所述紫檀烷類化合物的製備方法為:將原料苯酚類化合物在高價碘氧化劑(PIDA)和甲醇的作用下,合成得到反應底物醌單縮醛(QMA) 1l;再將2mmol醌單縮醛和親核試劑2H-苯並吡喃-7-基乙酸酯6a的混合物溶解在2.5ml的二氯甲烷(DCM)中,然後加入2.5ml六氟異丙醇(HFIP)和1mmol五氟苯甲酸(PFBA),在室溫下攪拌並用矽膠點板TLC監測測反應進程,待反應完成後再經過蒸發濃縮,然後通過快速矽膠色譜法(己烷/乙酸乙酯= 4/1)從反應濃縮液中直接進行產物的分離,最終得到產品紫檀烷6la。
優選的,所述具有高生物活性的天然活性藥物中間體的製備方法,包括所述吡咯烷基二氫吲哚類化合物的製備:將原料苯胺衍生物在高價碘氧化劑(PIDA)和甲醇的作用下,合成得到反應底物亞氨基醌縮醛1l;再將1-3mmol亞氨基醌縮醛和親核試劑1-甲苯磺醯基-2,3-二氫-1H-吡咯6b的混合物溶解在2-3ml二氯甲烷(DCM)中,然後依次加入2-3ml六氟異丙醇(HFIP)和0.5-1.5mmol五氟苯甲酸(PFBA),在室溫下攪拌並用矽膠點板TLC監測測反應進程,待反應完成後再經過蒸發濃縮,然後通過快速矽膠色譜法(己烷/乙酸乙酯= 4/1)從反應濃縮液中直接進行產物的分離,最終得到產品吡咯烷基二氫吲哚6ib。
進一步的,所述吡咯烷基二氫吲哚類化合物的製備方法為:將原料苯胺衍生物在高價碘氧化劑(PIDA)和甲醇的作用下,合成得到反應底物亞氨基醌縮醛1l;再將2mmol亞氨基醌縮醛和親核試劑1-甲苯磺醯基-2,3-二氫-1H-吡咯6b的混合物溶解在2.5ml二氯甲烷(DCM)中,然後依次加入2.5ml六氟異丙醇(HFIP)和1mmol五氟苯甲酸(PFBA),在室溫下攪拌並用矽膠點板TLC監測測反應進程,待反應完成後再經過蒸發濃縮,然後通過快速矽膠色譜法(己烷/乙酸乙酯= 4/1)從反應濃縮液中直接進行產物的分離,最終得到產品吡咯烷基二氫吲哚6ib。
優選的,所述具有高生物活性的天然活性藥物中間體的製備方法,包括所述反應底物的製備:將8-12mmol原料溶解在18-24ml甲醇中,得反應液,反應溫度控制在-2-2℃,再將8-12mmol高價碘氧化劑PhI(OAc)2溶解在等量的甲醇中,然後轉移至反應液中,逐漸升溫至室溫,保持該反應條件,持續攪拌8-12分鐘,充分反應完成後,加水稀釋,並用乙酸乙酯萃取,反應有機層用飽和碳酸氫鈉和飽和氯化鈉水溶液洗滌,用硫酸鈉乾燥,經真空濃縮後得到的反應物,通過矽膠柱色譜用己烷-乙酸乙酯純化,真空濃縮得到純的反應底物。
進一步的,所述反應底物的製備方法為:將10mmol原料溶解在20ml甲醇中,得反應液,反應溫度控制在0℃,再將10mmol高價碘氧化劑PhI(OAc)2溶解在20ml的甲醇中,然後轉移至反應液中,逐漸升溫至室溫,保持該反應條件,持續攪拌10分鐘,充分反應完成後,加水稀釋,並用乙酸乙酯萃取,反應有機層用飽和碳酸氫鈉和飽和氯化鈉水溶液洗滌,用硫酸鈉乾燥,經真空濃縮後得到的反應物,通過矽膠柱色譜用己烷-乙酸乙酯純化,真空濃縮得到純的反應底物。
將具有高生物活性的天然活性藥物中間體應用於抗真菌劑、抗微生物劑、抗蛇毒劑、抗腫瘤劑和抗HIV劑。進一步的,所述抗真菌劑、抗微生物劑、抗蛇毒劑、抗腫瘤劑和抗HIV劑為體內和/或體外藥劑。
本發明有益效果:
一、製備過程簡單、可靠、總收率高,其中紫檀烷類可達35%以上,吡咯烷基二氫吲哚類可達75%以上。
二、所用的DCM/六氟異丙醇HFIP溶劑不用進行乾燥處理,可直接使用市售溶劑,反應原料來源廣,在室溫下,使用五氟苯甲酸(PFBA),其反應中所利用的PFBA、DCM、HFIP也都能通過化學蒸餾法全部回收再利用,節約了生產成本,同時其能完全回收利用,使產品純度更高、安全綠色環保。
附圖說明
圖1為紫檀烷類化合物的反應路線圖。
圖2為吡咯烷基二氫吲哚類化合物的反應路線圖。
以上圖中所示僅代表其中一個流程。
具體實施方式
以下通過具體實施例的形式,對本發明的上述內容再做進一步的詳細說明。但不應將此理解為本發明上述主題的範圍僅限於以下的實施例。凡基於本發明上述內容所實現的技術均屬於本發明的範圍。
實施例1
醌單縮醛的製備
將12mmol對位取代苯酚溶解在22ml甲醇中,得反應液,反應溫度控制在1℃,再將10mmol高價碘氧化劑PhI(OAc)2溶解在22ml甲醇中,然後轉移至反應液中,逐漸升溫至室溫,保持該反應條件,持續攪拌10分鐘,充分反應完成後,加水稀釋,並用乙酸乙酯萃取,反應有機層用飽和碳酸氫鈉和飽和氯化鈉水溶液洗滌,用硫酸鈉乾燥,經真空濃縮後得到的反應物,通過矽膠柱色譜用己烷-乙酸乙酯純化,真空濃縮得到純的反應底物醌單縮醛1l,反應收率為92%。
實施例2
醌單縮醛的製備
將10mmol對位取代苯酚溶解在20ml甲醇中,得反應液,反應溫度控制在0℃,再將10mmol高價碘氧化劑PhI(OAc)2溶解在等量的甲醇中,然後轉移至反應液中,逐漸升溫至室溫,保持該反應條件,持續攪拌10分鐘,充分反應完成後,加水稀釋,並用乙酸乙酯萃取,反應有機層用飽和碳酸氫鈉和飽和氯化鈉水溶液洗滌,用硫酸鈉乾燥,經真空濃縮後得到的反應物,通過矽膠柱色譜用己烷-乙酸乙酯純化,真空濃縮得到純的反應底物醌單縮醛1l,反應收率為91%。
實施例3
亞氨基醌縮醛的製備
將12mmol對甲氧基-N-甲苯磺醯基苯胺溶解在20ml甲醇中,得反應液,反應溫度控制在2℃,再將10mmol高價碘氧化劑PhI(OAc)2溶解在22ml甲醇中,然後轉移至反應液中,逐漸升溫至室溫,保持該反應條件,持續攪拌8分鐘,充分反應完成後,加水稀釋,並用乙酸乙酯萃取,反應有機層用飽和碳酸氫鈉和飽和氯化鈉水溶液洗滌,用硫酸鈉乾燥,經真空濃縮後得到的反應物,通過矽膠柱色譜用己烷-乙酸乙酯純化,真空濃縮得到純的反應底物醌單縮醛1l,反應收率為93%。
實施例4
亞氨基醌縮醛的製備
將10mmol對甲氧基-N-甲苯磺醯基苯胺溶解在20ml甲醇中,得反應液,反應溫度控制在0℃,再將10mmol高價碘氧化劑PhI(OAc)2溶解在20ml甲醇中,然後轉移至反應液中,逐漸升溫至室溫,保持該反應條件,持續攪拌10分鐘,充分反應完成後,加水稀釋,並用乙酸乙酯萃取,反應有機層用飽和碳酸氫鈉和飽和氯化鈉水溶液洗滌,用硫酸鈉乾燥,經真空濃縮後得到的反應物,通過矽膠柱色譜用己烷-乙酸乙酯純化,真空濃縮得到純的反應底物醌單縮醛1l,反應收率為91%。
實施例5
紫檀烷類化合物的製備
將原料對位取代苯酚在高價碘氧化劑(PIDA)和甲醇的作用下,合成得到反應底物醌單縮醛(QMA) 1l;再將2.5mmol醌單縮醛和親核試劑2H-苯並吡喃-7-基乙酸酯6a的混合物溶解在2ml的二氯甲烷(DCM)中,然後加入2ml六氟異丙醇(HFIP)和1mmol五氟苯甲酸(PFBA),在室溫下攪拌並用矽膠點板TLC監測測反應進程,待反應完成後再經過蒸發濃縮,然後通過快速矽膠色譜法(己烷/乙酸乙酯= 4/1)從反應濃縮液中直接進行產物的分離,最終得到產品紫檀烷6la,總收率37%。
實施例6
紫檀烷類化合物的製備
將原料對位取代苯酚在高價碘氧化劑(PIDA)和甲醇的作用下,合成得到反應底物醌單縮醛(QMA) 1l;再將2mmol醌單縮醛和親核試劑2H-苯並吡喃-7-基乙酸酯6a的混合物溶解在2.5ml的二氯甲烷(DCM)中,然後加入2.5ml六氟異丙醇(HFIP)和1mmol五氟苯甲酸(PFBA),在室溫下攪拌並用矽膠點板TLC監測測反應進程,待反應完成後再經過蒸發濃縮,然後通過快速矽膠色譜法(己烷/乙酸乙酯= 4/1)從反應濃縮液中直接進行產物的分離,最終得到0.35mmol產品紫檀烷6la,總收率35%。
實施例7
吡咯烷基二氫吲哚類化合物的製備
將原料苯胺衍生物在高價碘氧化劑(PIDA)和甲醇的作用下,合成得到反應底物亞氨基醌縮醛1l;再將3mmol亞氨基醌縮醛和親核試劑1-甲苯磺醯基-2,3-二氫-1H-吡咯6b的混合物溶解在3ml二氯甲烷(DCM)中,然後依次加入2ml六氟異丙醇(HFIP)和0.5mmol五氟苯甲酸(PFBA),在室溫下攪拌並用矽膠點板TLC監測測反應進程,待反應完成後再經過蒸發濃縮,然後通過快速矽膠色譜法(己烷/乙酸乙酯= 4/1)從反應濃縮液中直接進行產物的分離,最終得到產品0.78mmol吡咯烷基二氫吲哚6ib,總收率78%。
實施例8
吡咯烷基二氫吲哚類化合物的製備
將原料苯胺衍生物在高價碘氧化劑(PIDA)和甲醇的作用下,合成得到反應底物亞氨基醌縮醛1l;再將2mmol亞氨基醌縮醛和親核試劑1-甲苯磺醯基-2,3-二氫-1H-吡咯6b的混合物溶解在2.5ml二氯甲烷(DCM)中,然後依次加入2.5ml六氟異丙醇(HFIP)和1mmol五氟苯甲酸(PFBA),在室溫下攪拌並用矽膠點板TLC監測測反應進程,待反應完成後再經過蒸發濃縮,然後通過快速矽膠色譜法(己烷/乙酸乙酯= 4/1)從反應濃縮液中直接進行產物的分離,最終得到產品0.75mmol吡咯烷基二氫吲哚6ib,總收率75%。
本發明製得的產品相關指標:
6a,12a-二氫-6H- [1,3]二氧雜環戊烯並[4',5':5,6]苯並呋喃並[3,2-c]苯並吡喃-3-基乙酸酯6la
淺黃色油狀物;1HNMR (400 MHz, CDCl3): = 2.39 (s, 3H), 3.95 (s, 3H), 3.98 (s, 3H), 4.03 (s, 3H), 6.92 (s, 1H),7.17(d, 1H, J = 7.3 Hz), 7.54-7.58 (m, 2H), 8.39 (s, 1H), 8.45 (d, 1H, J = 8.8 Hz) ppm。
5-甲氧基-1,8-二甲苯磺醯基-1,2,3,3a,8,8a-六氫吡咯並[2,3-b]吲哚6ib
淺黃色油狀物;1HNMR(400 MHz, CDCl3): = 2.39 (s, 3H), 3.95 (s, 3H), 3.98 (s, 3H), 4.03 (s, 3H), 6.92 (s, 1H), 7.17 (d, 1H, J = 7.3 Hz), 7.54-7.58 (m, 2H), 8.39 (s, 1H), 8.45 (d, 1H, J = 8.8 Hz) ppm。
顯然,根據本發明的上述內容,按照本領域的普通技術知識和手段,在不脫離本發明上述基本技術思想前提下,還可以做出其他多種形式的修改、替換或變更。