一種用於提高成骨細胞粘附和成骨性能的可降解醫用高分子三維材料的製備方法與流程
2023-08-08 09:53:26 2
本發明涉及一種可降解醫用高分子材料納米複合材料和仿生改性等技術領域,尤其涉及一種用於提高成骨細胞粘附和成骨性能的可降解醫用高分子三維材料的製備方法。
背景技術:
在口腔種植和牙周治療中,為獲得最佳的軟組織和硬組織的再生效果,引導骨再生(guidedboneregeneration,gbr)和引導組織再生現在已經成為一個標準的方法。引導骨組織再生技術是應用廣泛的口腔骨增量術。性能優越的gbr材料是保證gbr手術成功的基礎。因此,對gbr材料的研究成為口腔組織工程的重要組成部分。gbr技術主要是將屏障膜放置在骨缺損區,利用膜阻止生長較快的非成骨性細胞如上皮細胞等向缺損內長入,同時在膜下方維持一個空間,允許成骨性細胞優先遷移、生長,使骨再生、修復,最終在缺損區誘導形成新骨。在較大的骨缺損處需要同時充填骨替代材料,起到塑形和防止膜塌陷的作用。研究人員從各方面考慮薄膜的性能,通過幾種材料的複合或是對已經製備好的薄膜進行表面改性以及共混其他有利材料來滿足屏障膜引導骨再生的要求。它可通過一種主要的高分子材料與其它材料或者幾種高分子材料的納米複合,性能上揚長避短;或在膜基質中添加生物活性材料如生物活性陶瓷,生長因子,活性分子/蛋白,承載抗菌藥物,提高骨再生能力和抗感染能力;或改進位作工藝,優化調控薄膜孔隙結構,模擬天然細胞外基質,增強膜表面與細胞的相互作用。
在組織工程中,製備具有三維多孔結構的方法主要包括製備的三維結構材料具有多孔且孔結構互通等特點,利於成骨細胞的粘附和三維生長。靜電紡絲製造的組織再生膜具有模擬細胞外基質的納米纖維支架3d結構,在增強細胞-表面交互作用,誘導新骨形成上更具優勢。通過聚乳酸-羥基乙酸共聚物(plga)和可溶性蛋殼膜蛋白(solubleeggshellmembraneprotein,sep)按照不同比例混合製備出結構類似於ecm和天然蛋殼的電紡膜,plga大大改善了sep的機械性能,體外培養發現sep/plga納米纖維薄膜可明顯提高l-929成纖維細胞粘附和該細胞與纖維基質的交互作用。geistlichbio-gide®可吸收生物膜是由高純度的天然ⅰ型和ⅲ型膠原蛋白構成的生物膠原膜。它具有獨特的雙層結構設計,其緻密層可行使有效的屏障功能,對軟組織生長能起到導軌作用,膠原成份有促進傷口癒合的作用;疏鬆多孔層是三維膠原蛋白構成的框架結構,能夠促進細胞的長入同時還可起到穩定凝血塊的作用。
基於貽貝激發的仿生改性技術(mussel-inspiredtechnology)海洋貽貝通過足絲分泌粘附蛋白,蛋白中含有豐富的l-3,4-二羥基苯丙氨酸(dopa)具有超強粘附能力,利用與dopa結構相似的多巴胺模擬貽貝的粘附特性成為仿生學研究的熱點。多巴胺特有的粘附特性,能在基質表面沉積聚多巴胺塗層,直接將多種活性分子如蛋白、寡核苷酸、多糖、金屬離子、納米羥基磷灰石等固定到生物大分子上,方法簡單、反應條件溫和、綠色環保。研究證明gbr膜材料的表面多巴改性不影響細胞凋亡,還明顯提高了骨髓間充質幹細胞的粘附和成骨分化。
促進成骨性能的材料一直是研究的熱點。專利cn201610865987.2公開了生物陶瓷支架表面形成的聚多巴胺/ca-p納米層具有微納米結構,能夠促進骨間充質幹細胞在支架表面的粘附、增殖及分化;上海交通大學醫學院附屬第九人民醫院的專利cn201610665925.7採用靜電紡絲技術製備具有促進牙周膜細胞粘附和骨膜細胞粘附的材料並結合兼具抗菌活性的藥物,具有良好的生物相容性、骨修復和抗菌功能;專利cn201510300717.2公開了一種具有二級三維結構的複合骨修復材料及其製備方法,其複合材料綜合了納米羥基磷灰石、絲素和膠原三者的優點;專利cn201610963191.0披露了一種3d列印的聚己內酯/牡蠣殼粉複合材料及製備方法與應用和基於其的骨組織工程支架;北京科技大學的專利cn201610346507.1涉及一種牙科種植體利用聚多巴胺沉積羥基磷灰石,獲得一種具有優良生物活性的牙科種植體複合表面。綜上所述,到目前為止,仍然未見一種材料結合混合改性和表面改性,能夠獲得成骨活性物質在表面短時間的高表達,在體相的長時間的低表達的相關材料或者技術出現。
技術實現要素:
本發明的目的在於提供一種解決上述技術問題的用於提高成骨細胞粘附和成骨性能的可降解醫用高分子三維材料的製備方法。
為實現上述發明目的,本發明的一種用於提高成骨細胞粘附和成骨性能的可降解醫用高分子三維材料的製備方法,該方法包括如下步驟:
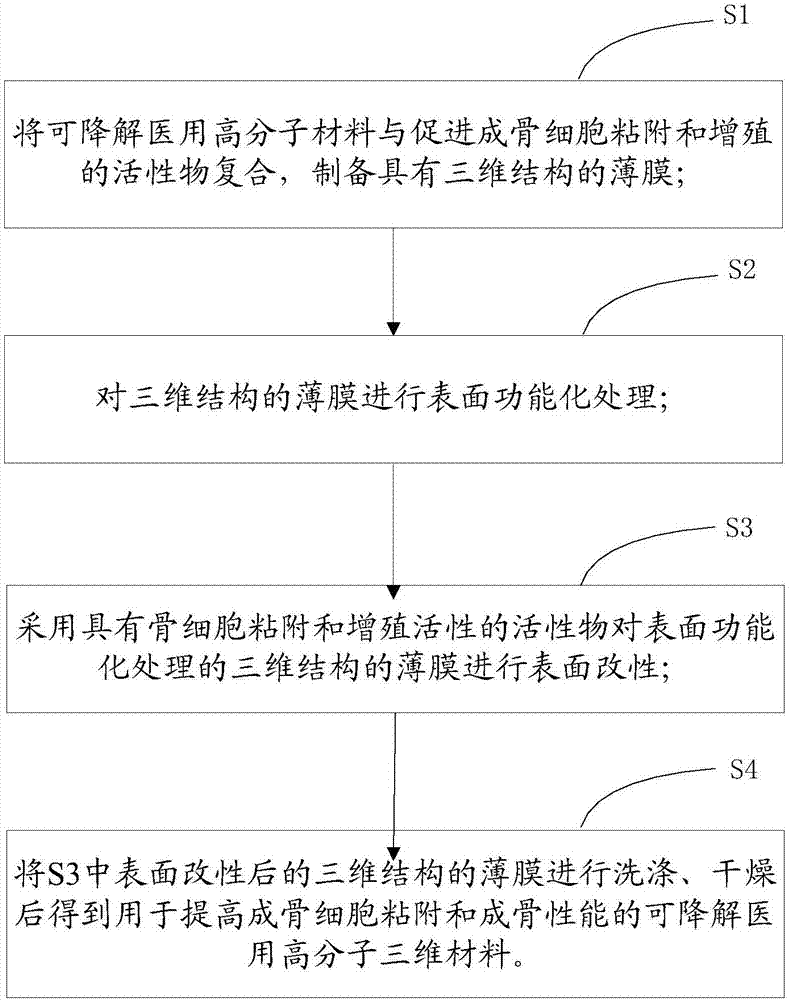
s1、將可降解醫用高分子材料與促進成骨細胞粘附和增殖的活性物複合,製備具有三維結構的薄膜;
s2、對三維結構的薄膜進行表面功能化處理;
s3、採用具有骨細胞粘附和增殖活性的活性物對表面功能化處理的三維結構的薄膜進行表面改性;
s4、將s3中表面改性後的三維結構的薄膜進行洗滌、乾燥後得到用於提高成骨細胞粘附和成骨性能的可降解醫用高分子三維材料。
作為本發明的進一步改進,所述可降解醫用高分子材料包括醫用級的脂肪族聚酯,聚乳酸、聚己內酯、聚乳酸-羥基乙酸共聚物、殼聚糖、纖維素、聚乙烯醇的一種或者幾種的組合,所述促進成骨細胞粘附和增殖的活性物包括膠原、明膠,多肽、蛋白、生長因子、殼聚糖、納米羥基磷灰石和磷酸三鈣一種或者幾種的組合。
作為本發明的進一步改進,所述三維結構的薄膜中,所述促進成骨細胞粘附和增殖的活性物的含量為0.05wt%~20wt%。
作為本發明的進一步改進,在步驟s1中,採用靜電紡絲法將可降解醫用高分子材料與促進成骨細胞粘附和增殖的活性物複合,製備具有三維結構的薄膜。
作為本發明的進一步改進,所述步驟s2中「對三維結構的薄膜進行表面功能化處理」具體為:
將三維結構的薄膜放入ph值為7.5~9.0,濃度為0.1wt%~2wt%的多巴胺tris-hcl溶液中,反應1-48h。
作為本發明的進一步改進,所述步驟s3中「採用具有骨細胞粘附和增殖活性的活性物對表面功能化處理的三維結構的薄膜進行表面改性」的反應條件為:具有骨細胞粘附和增殖活性的活性物的濃度不高於10wt%,反應的溫度為0~60℃,反應時間少於48h。
作為本發明的進一步改進,所述步驟s2還包括:對三維結構的薄膜進行表面功能化處理之前,對步驟s1中製備的清洗三維結構的薄膜進行清洗、除溶劑、乾燥處理。
與現有技術相比,本發明的有益效果是:
1、本發明提供的具有三維結構和強成骨活性的薄膜材料,其製備的關鍵點是成骨活性物質通過表面改性和混合改性的方法,在表面有高表達和體相長久低表達。成骨細胞的粘附是決定其後續增值和成骨性能的第一步,所以表面的膠原、生長因子等的高表達,能夠使得成骨細胞在材料表面短時間內吸附和生長。通過混合改性的方法材料具有表製備含有成骨活性物的薄膜,隨著聚合物的降解,活性物逐步釋放,實現良好的成骨性能;同時該方法極大的減少活性物質混合對材料力學的影響。
2、採用聚多巴胺的改性方法,適用性強,對反應的材料要求低;過程簡單可控,穩定性好等特點。此外其單體多巴胺無毒性,反應過程沒有有機溶劑。這些特點均為規模化製備奠定了基礎。此外電紡等製備三維結構的技術,均屬於成熟技術。因此,製備方法和技術便於推廣和在工業大生產中實現;
3、本發明可以有效控制成本。引導骨再生膜是高附加值產品,但是具有成骨細胞活性的物質其成本也非常高,例如骨生長因子。因此,通過表面改性的方法,可以減少活性物的使用,降低成本。
附圖說明
圖1為用於提高成骨細胞粘附和成骨性能的可降解醫用高分子三維材料的製備方法的步驟流程圖。
具體實施方式
下面結合實施例對本發明進行詳細的描述,但這些實施例並不限制本發明,本領域的普通技術人員根據實施例所作出等效變換或等效替代均包含在本發明的保護範圍內。
參圖1所示,本發明一實施例中用於提高成骨細胞粘附和成骨性能的可降解醫用高分子三維材料的製備方法,包括以下步驟:
s1、將可降解醫用高分子材料與促進成骨細胞粘附和增殖的活性物複合,製備具有三維結構的薄膜。具體地,是採用靜電紡絲法將可降解醫用高分子材料與促進成骨細胞粘附和增殖的活性物複合,製備具有三維結構的薄膜。在三維結構的薄膜中,所述促進成骨細胞粘附和增殖的活性物的含量為0.05wt%~20wt%,並且,添加的促進成骨細胞粘附和增殖的活性物均勻分布,不破壞高分子薄膜的結構和力學性能。由此製備的具有三維結構的薄膜材料具有多孔且孔結構互通等特點,利於成骨細胞的粘附和三維生長。當然,在本發明的其他實施例中,也可以利用3d列印和制孔法同樣能夠獲得相同特徵的三維結構的薄膜。
優選地,可降解醫用高分子材料包括醫用級的脂肪族聚酯,聚乳酸、聚己內酯、聚乳酸-羥基乙酸共聚物、殼聚糖、纖維素、聚乙烯醇的一種或者幾種的組合,所述促進成骨細胞粘附和增殖的活性物包括膠原、明膠,多肽、蛋白、生長因子、殼聚糖、納米羥基磷灰石和磷酸三鈣一種或者幾種的組合。
s2、對三維結構的薄膜進行表面功能化處理。先對步驟s1中製備的清洗三維結構的薄膜進行清洗、除溶劑、乾燥處理,再對三維結構的薄膜進行表面功能化處理。具體地,將三維結構的薄膜放入ph值為7.5~9.0,濃度為0.1wt%~2wt%的多巴胺tris-hcl溶液中,反應1-48h,使得多巴胺在三維結構的薄膜表面發生自聚,實現對三維結構的薄膜進行表面功能化。
s3、採用具有骨細胞粘附和增殖活性的活性物對表面功能化處理的三維結構的薄膜進行表面改性,反應條件為:具有骨細胞粘附和增殖活性的活性物的濃度不高於10wt%,反應的溫度為0~60℃,反應時間少於48h。優選地,具有骨細胞粘附和增殖活性的活性物包括膠原、生長因子、多肽、殼聚糖和明膠等。需要說明的是,對於不富含氨基的活性物(如i型膠原)來講,可以先通過低濃度戊二醛或乙二醛等預處理,提高其表面改性的效果。
s4、將s3中表面改性後的三維結構的薄膜進行洗滌、乾燥後得到用於提高成骨細胞粘附和成骨性能的可降解醫用高分子三維材料,通過上述方法製備的用於提高成骨細胞粘附和成骨性能的可降解醫用高分子三維材料能夠保持力學性能和結構,通過混合改性和表面修飾方法能夠實現表面短時高濃度和整體長久低表達成骨活性物的結合,明顯提高成骨細胞的粘附和三維生長,同時對材料的力學性能影響較小。
以下結合具體實施例來對本發明做進一步說明。
實施例一
稱取1gplga、10%殼聚糖,放入三氟乙醇溶液中,磁力攪拌,溶解均勻。在參數為正壓18.5kv,負壓2.8kv,溼度35%,溫度19℃下通過靜電紡絲方法製備具有三維結構的薄膜,將所得薄膜置於真空乾燥箱中乾燥48h,去除溶劑;將獲得的具有三維結構的薄膜材料進行多巴胺表面功能化處理,在ph值為8.5,濃度為0.3wt%多巴胺tris-hcl溶液中,反應24h後用去離子水洗滌3次。在2%的戊二醛中預處理1h後用去離子水洗滌3次。在5wt%的膠原溶液中,反應24h,控制溫度為37℃,最後用去離子水洗滌3次、放入真空乾燥箱中乾燥48h得到產品。
製備的電紡膜材料具有三維結構,前成骨細胞mc3t3-e1培養結果顯示,具有良好成骨性能。
實施例二:
稱取1gplga在三氟乙醇溶液中,磁力攪拌,溶解均勻,加入250μl膠原並磁力攪拌混勻。在參數為正壓16.23kv,負壓2.33kv,溼度32%,溫度19℃下通過靜電紡絲方法製備具有三維結構的薄膜,將所得薄膜置於真空乾燥箱中乾燥48h,去除溶劑;將獲得的具有三維結構的薄膜材料進行表面功能化處理,在ph值為8.5,濃度為0.1wt%多巴胺tris-hcl溶液中,反應24h後用去離子水洗滌3次。在6wt%的bmp2溶液中,反應36h,控制溫度為37℃,最後用去離子水洗滌3次、放入真空乾燥箱中乾燥48h,得到產品。
製備的電紡膜材料具有三維結構,前成骨細胞mc3t3-e1培養結果顯示,具有良好成骨性能。
實施例三:
稱取0.5gplga、0.5gpcl(聚己內酯)在三氟乙醇溶液中,超聲溶解均勻,加入250μl膠原並磁力攪拌均勻,再加0.03gβ-tcmp並攪拌均勻。在參數正壓16.85kv,負壓2.47kv,溼度30%,溫度18℃下通過靜電紡絲方法製備具有三維結構的薄膜,將製備的薄膜置於真空乾燥箱中乾燥48h,去除溶劑;將獲得的具有三維結構的薄膜材料進行表面功能化處理,在ph值為8.5,濃度為0.1wt%多巴胺tris-hcl溶液中,反應24h後用去離子水洗滌3次。在5wt%的膠原溶液中,反應24h,控制溫度為37℃,最後用去離子水洗滌3次、放入真空乾燥箱中乾燥48h,得到產品。
製備的電紡膜材料具有三維結構,前成骨細胞mc3t3-e1培養結果顯示,具有良好成骨性能。
實施例四:
稱取定量的plga、25%聚碳酸丁二醇酯先溶解均勻,加入10%殼聚糖並攪拌均勻。在參數正壓19.55kv,負壓1.23kv,溼度40%,溫度18℃下通過靜電紡絲方法製備具有三維結構的薄膜,將製備的薄膜置於真空乾燥箱中乾燥48h,去除溶劑;將獲得的具有三維結構的薄膜材料進行表面功能化處理,在ph值為8.5,濃度為0.2wt%多巴胺tris-hcl溶液中,反應24h後用去離子水洗滌3次。在5wt%的膠原溶液中,反應24h,控制溫度為37℃,最後用去離子水洗滌3次、放入真空乾燥箱中乾燥48h,得到產品。
製備的電紡膜材料具有三維結構,前成骨細胞mc3t3-e1培養結果顯示,具有良好成骨性能。
實施例五:
稱取0.5gplga、0.5gpcl在三氟乙醇溶液中,磁力攪拌,溶解均勻,加入10%納米羥基磷灰石並攪拌均勻。在參數正壓18.23kv,負壓2.5kv,溼度35%,溫度18℃下通過靜電紡絲方法製備具有三維結構的薄膜,將製備的薄膜置於真空乾燥箱中乾燥48h,去除溶劑;將獲得的具有三維結構的薄膜材料進行表面功能化處理,在ph值為8.5,濃度為0.3wt%多巴胺tris-hcl溶液中,反應24h後用去離子水洗滌3次。在4wt%的rgd多肽溶液中,反應36h,控制溫度為4℃,最後用去離子水洗滌3次、放入真空乾燥箱中乾燥48h,得到產品。
製備的電紡膜材料具有三維結構,前成骨細胞mc3t3-e1培養結果顯示,具有良好成骨性能。
實施例六:
稱取0.5gpcl、0.5g聚乙烯醇在三氟乙醇中溶解均勻,在正壓17.56kv,溼度33%,溫度18℃下通過靜電紡絲方法製備具有三維結構的薄膜,將製備的薄膜置於真空乾燥箱中乾燥48h,去除溶劑;將獲得的具有三維結構的薄膜材料進行表面功能化處理,在ph值為8.5,濃度為0.3wt%多巴胺tris-hcl溶液中,反應24h後用去離子水洗滌3次。在8wt%的羧酸化殼聚糖溶液中,反應24h,控制溫度為45℃,最後用去離子水洗滌3次、放入真空乾燥箱中乾燥48h,得到產品。
上文所列出的一系列的詳細說明僅僅是針對本發明的可行性實施方式的具體說明,它們並非用以限制本發明的保護範圍,凡未脫離本發明技藝精神所作的等效實施方式或變更均應包含在本發明的保護範圍之內。