具有聚集誘導發光性質的螢光探針及其製備方法和用途與流程
2023-08-12 22:32:36 1
本發明屬於表面活性劑檢測領域,特別涉及一種陰離子表面活性劑的檢測方法。
背景技術:
陰離子表面活性劑,尤其是烷基磺酸鹽是商品中最重要的表面活性劑並被大量生產,如:十二烷基苯磺酸鈉(sdbs),月桂基硫酸鈉(sls),十二烷基硫酸鈉(sds)等。它們在清潔、農業、化妝品和醫藥工業被廣泛用作洗滌劑、乳化劑、起泡劑和分散劑。由於它們的應用廣泛,其在水體中的殘餘物對生態環境可能造成嚴重危害,如:抑制水生物活性,加速油質汙染物的擴散。此外,陰離子表面活性劑可能對人眼和皮膚造成刺激並引起發炎。為了使陰離子表面活性劑的檢測甚至在家中或者偏遠地區也可實施,非常需要發展一種操作簡單、成本低廉的有效的檢測方法。目前,在文獻中,已經報導了大量的用於檢測陰離子表面活性劑的方法,如gc/lc-ms,比色法、電勢測定法、免疫分析法、毛細管電泳和流動注射分析法。然而,這些方法還存在著以下缺點,如:需要大型固定儀器、操作繁瑣、使用有毒的氯代溶劑和難以原位分析。
近年來,螢光法檢測陰離子表面活性劑顯示出了顯著的優點,如:靈敏度高、操作簡單成本低。然而,具有聚集誘導螢光淬滅(acq)性質的螢光材料在聚集態下,容易發生螢光的自淬滅,而且其自身在溶液態的高背景螢光也會明顯降低檢測過程中的信噪比。
與聚集誘導螢光淬滅(acq)相反,具有聚集誘導發光(aie)性質的螢光探針,在稀溶液中發光很弱,但在聚集態發光很強。通過形成發光聚集體,aie探針已被廣泛用於檢測金屬離子、酶、dna等。此外,aie分子還被用於檢測臨界膠束濃度、監測膠束包裹的藥物釋放、利用螢光成像監測膠束的形態轉變等過程。
技術實現要素:
:
本發明的目的是提供一種具有聚集誘導發光(aie)性質的螢光探針,其可以用於檢測陰離子表面活性劑。
本發明的又一目的是提供上述螢光探針的製備方法以及其用途。
本發明目的基於如下技術方案實現:
一種式i所示的化合物:
其中:
r1,r2獨立的為氫、c1-18烷基、滷素、c1-18烷基氧基、c1-18烷基硫基、芳基、雜芳基;r3,r4,r5獨立的為c1-30烷基;r6為直鍵、c1-6亞烷基;a-為陰離子。
所述的烷基可為直鏈或支鏈烷基;例如,甲基、乙基、丙基、丁基、異丁基、叔丁基等。
所述的芳基指具有6-20個碳原子的單環或多環芳族基團,代表性的芳基包括:苯基、萘基、蒽基、芘基等。
所述的雜芳基指具有1-20個碳原子、1-4個選自n、s、o雜原子的單環或多環雜芳族基團,代表性的雜芳基包括:吡咯基、吡啶基、嘧啶基、咪唑基、噻唑基、吲哚基、氮雜萘基、氮雜蒽基、氮雜芘基等。
在一個實施方式中,r1,r2獨立的為氫、c1-6烷基、滷素、c1-6烷基氧基、c1-6烷基硫基。
在另一個實施方式中,r3,r4,r5中至少一個為c8-30烷基,更優選為c10-24烷基,又優選為c12-22烷基,或c14-20烷基。
在又一個實施方式中,r3,r4,r5中的一個基團為長鏈烷基,而另兩個基團為短鏈烷基,例如,r3,r4,r5中的一個基團為c8-30烷基,更優選為c10-24 烷基,又優選為c12-22烷基,或c14-20烷基;而另兩個基團獨立的為c1-8烷基,更優選為c1-6烷基。
在又一個實施方式中,r6為直鍵、-ch2-、-ch2ch2-、-ch2ch2ch2-。
優選的,所述陰離子為滷離子、高氯酸根離子、硫酸根離子、硝酸根離子、六氟磷酸根離子等。所述滷離子優選為氯離子。
優選的,所述通式i為如下通式(ia):
其中,r1、r2、r3、r4、r5、a-如上述所定義。
優選的,式ia中,r1為氫,r2為氫,r3,r4為甲基,r5為十八烷基,a-為陰離子。
更優選,式i化合物選自:
n-(3-(苯並[d]噻唑-2-基)-4-羥基苄基)-n,n-二甲基十八烷基-1-氯化銨;
本發明還提供了一種上述式i化合物的製備方法,包括將式ii化合物與式iii化合物在質子酸催化和氧化試劑的氧化下,反應得到式i化合物:
其中,r1、r2、r3、r4、r5、r6、a-如上述所定義。
優選的,所述質子酸為濃鹽酸,所述氧化試劑是雙氧水。
所述式ii化合物可由如下製備方法獲得,包括:將式iv化合物與式v化合物反應得到式ii化合物。
其中,r2、r3、r4、r5、r6、a-如上述所定義。
本發明所述的式i化合物具有聚集誘導發光(aie)性質。所述結構中包括2-(2'-羥基苯基)苯並噻唑(hbt)分子骨架,其具有分子內六元環氫鍵和連接苯並噻唑單元和取代苯單元的c–n單鍵。由於其激發態質子分子內轉移過程(esipt)性質,具有醇式和酮式發射,分別具有正常的斯託克位移和很大的斯託克位移(大於150nm)。由於分子內氫鍵容易受到極性溶劑的破壞,醇式和酮式的發射比例與分子所處環境的極性高度相關。在極性溶劑中,所述化合物僅表現為醇式發光。而在聚集態時,分子結構之間會緊密排列,抑制分子內c–n單鍵的旋轉,且會保護分子內氫鍵免受外界環境的幹擾。由於所述化合物的螢光發射對環境的極性和分子的聚集態非常敏感,因此它們特別適於作為表面活性劑的傳感材料。
相對於單一的陽離子或陰離子表面活性劑體系,即使在遠低於各自臨界膠束濃度的情況下,具有相反電荷的表面活性劑的混合體系在溶液中亦傾向於形成陰陽離子聚集體或膠束,這是由於靜電和疏水的協同作用會有效降低表面張力造成的。本發明基於aie染料在聚集態高效發光的特性,利用具有aie性質的帶正電的兩親性探針分子(式i化合物),通過原位形成陰陽離子聚集體或膠束來檢測陰離子表面活性劑。通過在聚集體中分子間的緊密排列,hbt骨架的分子內運動被限制且分子內氫鍵被保護以防止環境幹擾(見圖1)。
進而,本發明還提供了所述式i化合物在檢測陰離子表面活性劑中的用途。
根據本發明,所述的陰離子表面活性劑可為羧酸鹽、硫酸酯鹽、磺酸鹽和磷酸酯鹽。優選的,所述的陰離子表面活性劑為連接有磺酸基團的陰離子表面活性劑,例如十二烷基苯磺酸鈉(sdbs),十二烷基硫酸鈉(sds)等。
本發明所述的化合物與陰離子表面活性劑形成陰陽離子聚集體。在極性溶 劑中,當本發明所述化合物與陰離子表面活性劑形成的陰陽離子膠束的濃度高於其臨界膠束濃度時,本發明所述化合物與陰離子表面活性劑會形成陰陽離子膠束。使得本發明所述的化合物所處的微環境極性變小。從而保護了所述化合物分子內的氫鍵。所述的混合體系中陰陽離子膠束的臨界濃度顯著低於陰離子表面活性劑的臨界膠束濃度。
本發明所述的化合物可用於監測陰陽離子膠束的形成過程,具有高效且靈敏的優點。本發明所述的化合物針對陰離子表面活性劑的檢測限(3δ/s)小於0.08μm,遠低於基於亞甲基藍分光光度法的國家標準(檢測限約為0.14μm)。
此外,基於激發態分子內質子轉移(esipt)和分子內運動受限(rim)機制,在陰離子表面活性劑濃度較低時,式i化合物的酮式/烯醇式的發光比率與陰離子表面活性劑的濃度成線性關係,因此,可以通過式i化合物的酮式/烯醇式的發光比率來定量確定水中表面活性劑的濃度。
本發明還提供了一種檢測陰離子表面活性劑的方法,包括,在陰離子表面活性劑的溶液中加入本發明所述的式i化合物,在螢光下觀察或檢測螢光光譜。
本發明所述的化合物在水溶液中具有微弱的螢光發射,但當其與陰離子表面活性劑結合生成聚集體後會發出很強的螢光,因此,它可以用作優異的點亮探針,實現對負電荷外膜包圍的細菌的免洗螢光成像。
進一步的,本發明提供了式i化合物在細菌免洗螢光染色的用途。
根據本發明,所述的細菌為具有負電荷外膜包圍的細菌,例如大腸桿菌、枯草桿菌、金黃色葡萄球菌等。
本發明所述的式i化合物可被用於直接染色細菌而無需進一步洗滌步驟。並且表現出非常低的針對細菌的毒性,有利於其針對細菌的螢光染色實驗。
由於細菌的外膜是由帶負電荷的兩親性分子組成的,而本發明所述的式i化合物是帶有正電荷的兩親性分子,因此其可以通過靜電及疏水作用與細菌外膜形成強的親和力;與之結合後,所述化合物的轉動等分子內運動會受到限制,根據聚集誘導發光的機制,分子內運動受限後,其發光會明顯增強。而溶解在溶液中的未與細菌結合的所述化合物,由於分子內運動,導致發光很弱。可見,本發明所述的化合物與細菌結合後發強螢光,而在溶液中發光微弱,在該高對比度下,僅觀察到細菌上的螢光信號。而傳統的螢光材料,在稀溶液中發光很 強,因此需要離心洗滌,除去稀溶液中的螢光分子,才可以用於觀察細菌上的螢光信號。
本發明還提供一種細菌免洗螢光染色的方法,包括,將本發明所述的式i化合物與細菌共培養,無需分離直接觀察其螢光,例如使用雷射掃描共焦顯微鏡進行觀察。
本發明至少具有以下有益效果:
1、本發明所述的化合物針對陰離子表面活性劑的檢測限(3δ/s)小於0.8μm,遠低於基於亞甲基藍分光光度法的國家標準,且不需要氯仿等有毒試劑的萃取、濃縮,可以直接原位進行檢測,操作十分方便。
2、本發明所述的化合物可以用作優異的點亮探針,能夠對細菌進行免洗螢光成像,無需分離。
3、本發明所述化合物製備簡單。
附圖說明
圖1為加入sdbs之前和之後的hbt-c18的假設的堆積模型。具有aie性質的螢光探針hbt-c18通過形成膠束點亮檢測陰離子表面活性劑。
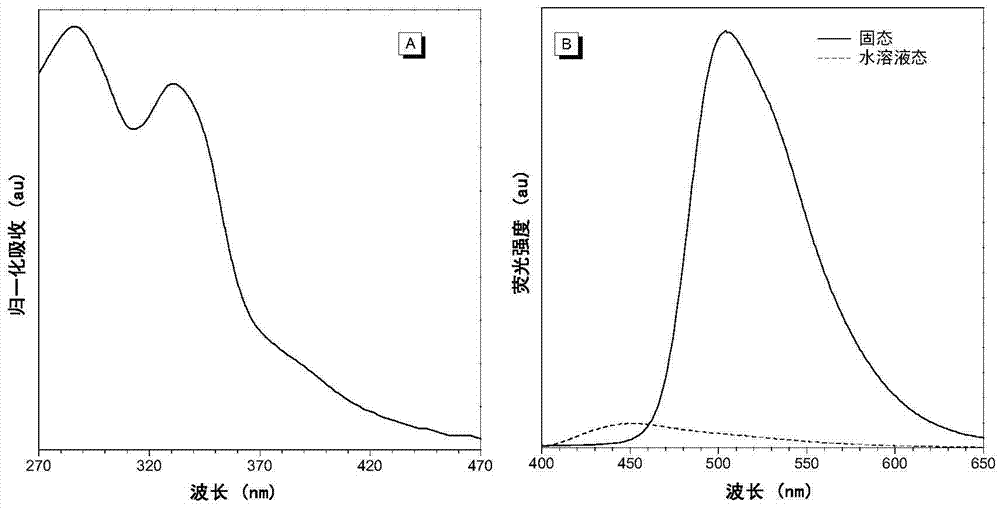
圖2(a)為hbt-c18在h2o/dmso(99:1,v/v)的溶液中的歸一化的紫外吸收光譜;(b)為hbt-c18在h2o/dmso(99:1,v/v)的溶液中及固態下的螢光發射光譜。[hbt-c18]=5μm;λex=334nm。
圖3(a)為隨著sdbs濃度的增加hbt-c18(5μm)在h2o/dmso(99:1,v/v)的溶液中的螢光發射譜圖;(b)為隨著sdbs濃度的增加,hbt-c18在510nm和450nm處的螢光發射強度變化;(c)為隨著sdbs濃度的增加,hbt-c18在510nm和450nm處的螢光發射強度的比例(i510/i450)變化。λex=334nm.
圖4為hbt-c18(5μm)在h2o/dmso(99:1,v/v)中隨sdbs濃度變化的螢光量子產率。
圖5為hbt-c18在不同sdbs濃度下(0,2.0,8.0μm)的(a)歸一化紫外吸收和(b)螢光發射光譜圖。[hbt-c18]=5μm;λex=334nm.
圖6為hbt-c18在510nm和450nm處的螢光發射強度的比例(i510/i450)隨sdbs濃度的變化。λex=334nm.內部為線性回歸方程。
圖7(a)為hbt-c18在510nm和450nm處的(i510/i450)螢光發射強度的比例對sdbs濃度的對數作圖;(b)為通過動態光散射測得的膠束粒徑,pdi=0.434;(c)、(d)為hbt-c18(5μm)/sdbs(8μm)在h2o/dmso(99:1,v/v)溶液中形成的膠束的(c)sem和(d)tem圖。
圖8為在不同表面活性劑及離子的存在下,hbt-c18在510nm和450nm處的螢光發射強度的比例(i510/i450)變化。
圖9為明場和螢光下的大腸桿菌的染色照片。(hbt-c18=10μm).比例尺=10μm.
圖10為e.coli細菌在不同濃度hbt-c18(0,10,20,40,60,80和100μm)下的存活率圖。
具體實施方式
下面結合具體實施例和說明書附圖,對本發明作進一步闡述。
實施例1
按照如下合成路線,具體合成以下化合物:
(1)n-(3-甲醯基-4-羥基苄基)-n,n-二甲基十八烷基-1-氯化銨(化合物3)的合成
在回流條件下將5-(氯甲基)-2-羥基水楊醛(340mg,2mmol)和n,n-二甲基十八烷-1-胺(594mg,2mmol)的混合物攪拌5小時。在反應完成後,過濾出沉澱並用乙醚洗滌(10ml×2),在真空下乾燥得到白色固體產物化合物3,產率為90%(841mg).
1hnmr(cdcl3,500mhz):δ11.26(brs,1h),8.16(d,j=3.5hz,1h),7.80(d,j=8.5hz,1h),7.09(d,j=8.5hz,1h),5.21(d,j=8.5hz,2h),3.47(t,j=8.5hz,2h),3.27(s,6h),1.26–1.25(m,32h),0.88(t,j=7.0hz,3h);13cnmr(d6-dmso,125mhz):190.0,162.3,140.1,132.9,122.5,118.8,118.0,65.3,63.1, 48.7,31.3,29.0,29.0,28.9,28.76,28.66,28.5,25.8,22.1,21.7,13.9;hrms(esi):m/z[m-cl]+計算值為c28h50no2:432.3836;實際測量值為:432.3845.
(2)n-(3-(苯並[d]噻唑-2-基)-4-羥基苄基)-n,n-二甲基十八烷基-1-氯化銨(hbt-c18)的合成
在n-(3-甲醯基-4-羥基苄基)-n,n-二甲基十八烷基-1-氯化銨(467mg,1.0mmol)和2-氨基苯硫酚(150mg,1.2mmol)的甲醇溶液中加入濃hcl(37wt.%,100mg,1.0mmol),所得混合液在室溫下攪拌10分鐘。隨後在混合液中加入h2o2(30wt.%,113mg,1.0mmol),並進一步在室溫下攪拌2小時。在反應完成後,蒸發掉溶劑,所得剩餘物用二氯甲烷/正己烷重結晶,得到白色固體產物n-(3-(苯並[d]噻唑-2-基)-4-羥基苄基)-n,n-二甲基十八烷基-1-氯化銨(hbt-c18)(120mg,產率21%)
1hnmr(cdcl3,500mhz):δ12.87(brs,1h),8.28(s,1h),7.97(d,j=8.0hz,1h),7.85(d,j=7.5hz,1h),7.63(d,j=8.0hz,1h),7.49(t,j=8.0hz,1h),7.40(t,j=8.0hz,1h),7.15(d,j=7.0hz,1h),5.17(s,2h),3.47(t,j=7.5hz,2h),3.31(s,6h),1.26–1.23(m,32h),0.88(t,j=7.0hz,3h);
13cnmr(cdcl3,125mhz):168.2,159.7,136.8,133.7,132.7,126.9,126.0,122.1,121.8,118.8,118.2,117.3,67.0,63.8,49.6,31.9,29.69,29.65,29.6,29.4,29.4,29.3,26.4,22.9,22.7,14.1;hrms(esi):m/z[m-cl]+計算值為c34h53n2os:537.3873;實際測量值為:537.3880.
圖2(a)為hbt-c18在h2o/dmso(99:1,v/v)的溶液中的歸一化的紫外吸收光譜;圖2(b)為hbt-c18在h2o/dmso(99:1,v/v)的溶液中及固態下的螢光發射光譜。[hbt-c18]=5μm;λex=334nm.由圖中可以看出,在溶液態,僅觀察到在約450nm處的醇式發光。這是由於在水溶液中的環境幹擾造成的。固態粉末時,在510nm觀測到具有大的stokes位移(176nm)的強發光,源於激發態分子內質子轉移(esipt)過程,而且由於分子內氫鍵被保護且其自由運動受到抑制,因此它可以發射強的酮式發光。大的stokes位移(176nm)是激發態分子內質子轉移(esipt)過程的典型特徵。hbt-c18在h2o/dmso(體積比99:1)溶液中和固態下的量子產率分別為0.2%和50.5%,清楚地證實了其aie性質。
實施例2:陰離子表面活性劑的檢測
將15μl實施例1製備的hbt-c18的dmso(1.0mm)母液加入2.985ml含有不同含量陰離子表面活性劑十二烷基苯磺酸鈉(sdbs)的純水中,漩渦震蕩10s,用螢光分光計測定螢光(激發波長334nm)。
圖3(a)為隨著sdbs濃度的增加hbt-c18(5μm)在h2o/dmso(99:1,v/v)的溶液中的螢光發射譜圖;(b)為隨著sdbs濃度的增加,hbt-c18在510nm和450nm處的螢光發射強度變化;(c)為隨著sdbs濃度的增加,hbt-c18在510nm和450nm處的螢光發射強度的比例(i510/i450)變化。λex=334nm.
圖4為hbt-c18(5μm)在h2o/dmso(99:1,v/v)中隨sdbs濃度變化的螢光量子產率。由圖中可以看出,hbt-c18的螢光量子產率從0.2%增加至8.5%。這是由於形成了陰陽離子聚集體,而且分子內氫鍵被保護。
圖5為hbt-c18在不同sdbs濃度下(0,2.0,8.0μm)的(a)歸一化紫外吸收和(b)螢光發射光譜圖。[hbt-c18]=5μm;λex=334nm.由圖中可以看出,在加入sdbs後,在370nm~420nm的拖尾峰的吸收光譜消失,分子內氫鍵得到保護,發出強烈的酮式螢光。
圖6為hbt-c18在510nm和450nm處的螢光發射強度的比例(i510/i450)隨sdbs濃度的變化。由圖中可以看出在450nm和510nm檢測到的發光比值(i510/i450)與sdbs增加的量相比逐漸增加。λex=334nm.內部為線性回歸方程。基於信噪比的計算方法,針對sdbs的檢測限(3δ/s)為0.05μm,遠低於基於亞甲基藍分光光度法的國家標準(檢測限約為0.14μm)。
圖7(a)為hbt-c18在510nm和450nm處的(i510/i450)螢光發射強度的比例對sdbs濃度的對數作圖;如圖所示,在hbt-c18/sdbs混合體系中的陰陽離子膠束的臨界濃度為2.39μm,顯著低於sdbs的臨界膠束濃度(1.2mm)。
圖7(b)為通過動態光散射(dls)測得的膠束粒徑,pdi=0.434。由圖所示,膠束的直徑平均為223.8nm。
圖7(c)、(d)為hbt-c18(5μm)/sdbs(8μm)在h2o/dmso(99:1,v/v)溶液中形成的膠束的掃描電鏡(sem)和透射電鏡(tem)圖。圖中顯示膠束為 球形結構,其半徑為90~200nm,相對於dls的結果,測得的膠束直徑略小,這是由於在樣品製備過程中,由溶液態轉變為乾燥的狀態,溶液揮發會導致膠束的收縮造成的。
實施例3:
不同表面活性劑及離子的檢測
將15μl實施例1製備的hbt-c18的dmso(1.0mm)母液加入2.985ml含有不同表面活性劑及離子(十二烷基苯磺酸鈉(sdbs),十二烷基硫酸鈉(sds)、曲通x-100、溴化十六烷基三甲基銨(ctab)和簡單的離子(如:na+,k+,mg2+,ca2+,zn2+no3–,hco3–,hco32–,po43–,so42–))的純水中,漩渦震蕩10s,用螢光分光計測定螢光(激發波長334nm)。結果如圖8所示。
圖8為在不同表面活性劑及離子的存在下,hbt-c18在510nm和450nm處的螢光發射強度的比例(i510/i450)變化。該探針對連有磺酸基的陰離子表面活性劑有很高的靈敏性,包括:十二烷基苯磺酸鈉(sdbs)、十二烷基硫酸鈉(sds),而對中性的表面活性劑曲通x-100和陽離子表面活性劑溴化十六烷基三甲基銨(ctab),以及多種簡單的陽離子(如:na+,k+,mg2+,ca2+,zn2+)、陰離子(如:no3–,hco3–,hco32–,po43–,so42–)則響應很弱。
實施例4:細菌螢光染色
(1)細菌培養
大腸桿菌(e.coli,strainatcc15224)購買自vwrinternational,llc。將該細菌的單菌落接種在5ml的lb基質中,在37℃下震動(150rpm)。之後,將1ml細菌懸濁液接種到50ml的新鮮基質中,在37℃震動下培養5h,以實現對數中期生長。
(2)e.coli的螢光染色
在37℃下將e.coli培養12小時,通過pbs將細菌的濃度稀釋至107cfu/ml,隨後用10μm實施例1製備的hbt-c18在37℃下培養0.5小時。用雷射掃描共焦顯微鏡技術進行螢光成像實驗(leicatcs-sp8,germany),在405nm激發並用508–558nm的濾光片。結果如圖9所示。
圖9為明場和螢光下的大腸桿菌的染色照片。(hbt-c18=10μm).比例尺=10μm.由圖中可以看出,在雷射掃描共聚焦顯微鏡下,所示大腸桿菌的螢光圖 像清晰可見,背景噪音的影響可以忽略。因此hbt-c18探針可被用於直接染色細菌無需進一步分離、洗滌步驟。
(3)抗菌實驗
將不同濃度的實施例1製備的hbt-c18探針加入1ml細菌濃度為107cfu/ml的lb介質中,測定hbt-c18的抗菌活性。在37℃下震蕩(150rpm)培養2h,所得溶液用9mlpbs稀釋。用移液器粗略地混合後,將溶液轉移至15ml的試管中,進一步渦流振蕩混合細菌3min。隨後,該細菌溶液被稀釋100,101和102倍,隨後取10μl稀釋的細菌懸濁液,用瓊脂板評估細菌的活性。
圖10為e.coli細菌在不同濃度hbt-c18(0,10,20,40,60,80和100μm)下的存活率圖。從圖中可以看出,hbt-c18探針表現出非常低的針對細菌的毒性,有利於其針對細菌的螢光染色實驗。
儘管結合優選實施例對本發明進行了說明,但本發明並不局限於上述實施例,應理解,這些實施例僅用於說明本發明而不用於限制本發明的範圍。此外應理解,在閱讀了本發明講授的內容之後,本領域的技術人員可以對本發明作各種不背離本發明宗旨的改動或修改,這些等價形式同樣落於本申請所附權利要求書所限定的範圍。