一種兼具靶向性和螢光特性的pH敏感殼聚糖載藥膠束及其製備方法與流程
2023-07-26 00:09:17 1
本發明屬於本發明涉及化學及生物醫藥交叉領域,具體涉及一種兼具靶向性和螢光特性的pH敏感殼聚糖載藥膠束及其製備方法。
背景技術:
癌症是一類嚴重危害人類健康的重大疾病,引起了國內外研究學者的廣泛關注。在癌症治療方法中,採用抗癌藥進行化學治療是對癌症患者手術後徹底殺滅體內癌細胞的最有效方法之一。但抗癌藥在殺死癌細胞的同時,也對機體內的某些正常器官細胞同樣造成嚴重傷害,並且對某些組織器官如肝、腎、心、肺等也造成損害,嚴重影響了癌症患者的治癒效果和生活質量。基於此,如何提高抗癌藥輸運系統的靶向性一直以來是國內外研究學者關注的熱點。
理想的載藥系統除了能夠縮短藥物的體內半衰期,延長體內作用時間,使藥物被動靶向富集於病灶周圍,還應保證最大程度地被腫瘤細胞攝取並在被攝取後將藥物迅速釋放,這就要求載藥膠束系統能夠具有更準確的靶向性和定時定點釋放性。研究發現,人體內正常細胞的血液環境pH在7.4左右,而腫瘤組織細胞間質呈弱酸性。利用這一特性,研究者們構建了pH敏感型載藥膠束,這一載藥膠束可靶向作用於腫瘤組織並通過癌細胞的內化作用,經pH為5.5-6.0的內涵體及pH低至4.5-5.0的溶酶體轉運,實現靶向給藥和定點釋藥的目的。
隨著對腫瘤研究的不斷深入,研究者在實現抗癌藥的靶向輸運和定點釋放後,更多地開始關注抗癌藥與腫瘤細胞作用過程的動力學特徵及機制研究,以獲取更多腫瘤細胞繁殖和控制的關鍵因素。因此,抗癌藥與腫瘤細胞的可視化作用及信息的表達顯得越來越重要。在眾多的生物體信息傳感技術中,螢光技術在核酸標記、人體癌細胞的靶向特異性標記、腫瘤診斷、治療和藥物療效評價等過程中,表現出靈敏、快速、對機體無損傷檢測及原位傳感癌細胞狀態信息等優異特性。
基於上述兩點,抗癌藥的靶向輸運和其與腫瘤細胞作用過程的可視化及原位信息傳感的結合,對抗癌藥與癌細胞作用規律的揭示、提高療效和減少患者機體損傷均具有重要意義。
技術實現要素:
本發明所要解決的技術問題是,針對抗癌藥輸運系統的靶向性需求和抗癌藥與腫瘤癌細胞作用過程的可視化需求,提供一種兼具靶向性和螢光特性的pH敏感載藥殼聚糖膠束的製備方法,從而構建一種集靶向性、pH敏感性、隱形性和螢光示蹤等多功能於一體的殼聚糖載藥膠束。
為了解決上述技術問題,本發明採用的技術方案是:一種兼具靶向性和螢光的pH敏感殼聚糖載藥膠束的製備方法,該載藥殼聚糖膠束以殼聚糖為主體,通過還原胺化反應在其上接枝疏水的辛基鏈,在以酸敏感β-羧酸醯胺鍵為連接臂接枝親水的羧基環己甲醯基,從而構建得到兩親性pH敏感殼聚糖;然後,再以聚乙二醇為連接臂,分別在兩親性pH敏感殼聚糖兩端接枝葉酸和螢光染料噻唑橙;利用葉酸的靶向性、連接臂聚乙二醇的「隱形效應」和兩親性殼聚糖的pH敏感性,提高殼聚糖載藥膠束的靶向性;利用螢光染料噻唑橙的螢光特性,實現殼聚糖載藥膠束與腫瘤細胞作用過程可視化。
具體地說,包括以下步驟:
(1)N-辛基-N』-(2-羧基環己甲醯基)殼聚糖的合成
稱取1質量份的殼聚糖(CTS),並將其置於無水甲醇(MeOH)中,在30℃和機械攪條件下進行溶脹;而後向反應液中緩慢滴加2.07-2.48質量份的辛醛,保溫反應5-36h後,分六個批次加入硼氫化鈉0.5-1質量份的NaBH4,室溫下繼續還原12-36h,還原反應完成後調節反應液pH值至6-8;反應液抽濾,並用甲醇和去離子水交替洗滌濾餅,而後濾餅經真空乾燥後得到淡黃色的N-辛基殼聚糖(OC);
稱取1質量份的前述N-辛基殼聚糖(OC)並將其置於反應瓶中,採用無水二甲基亞碸(DMSO)進行溶脹,在室溫條件下向反應瓶中滴加1-2質量份的六氫苯酐(C8H10O3),升溫至80℃反應12-36h;反應結束後,將反應液倒入去離子水中並置於冰水浴中,並調節反應液pH值至9-10;反應液進行抽濾,將濾液置於分子量為3500的透析袋中進行透析,而後將透析袋中反應液經冷凍乾燥即可得黃白色絮狀固體N-辛基-N』-(2-羧基環己甲醯基)殼聚糖(OCCC);
(2)聚乙二醇的葉酸接枝物(FA-PEG-NH2)及噻唑橙接枝物(TO-PEG-NH2)的合成
以二甲基亞碸為溶劑,稱取1質量份的葉酸(FA),並以1.38-1.72質量份的N-羥基琥珀醯亞胺(NHS)和0.35-0.70質量份的二環己基碳二亞胺(DCC)為縮合劑,在室溫下避光攪拌4h,而後向反應體系中加入3.45-6.90質量份的端氨基聚乙二醇(NH2-PEG-NH2),繼續攪拌反應8-12h。反應結束後向體系加入去離子水,抽濾除去不溶物,所得濾液離心後進行冷凍乾燥;冷凍乾燥所得固體產物在氯仿中溶解並進行抽濾,濾液用乙醚進行重結晶,過濾所得析出物再次溶於去離子水中,並再次經冷凍乾燥得橘黃色海綿狀產物,即聚乙二醇的葉酸接枝物(FA-PEG-NH2);
採用同樣的合成方法,將前述葉酸(FA)換成1.38質量份的螢光染料噻唑橙(TO),其它反應條件不變,可得紅色海綿狀固體,即聚乙二醇的噻唑橙接枝物(TO-PEG-NH2);
(3)聚乙二醇的丁二酸酐(SA)接枝物的合成
稱取1質量份的步驟(2)中所製備的FA-PEG-NH2並將其溶於無水二甲基亞碸(DMSO)中,加入0.2-0.3質量份的丁二酸酐(SA),室溫下攪拌反應,反應結束後向反應液加入0.75-1.5質量份的氯仿,並將反應液滴入冷乙醚中析出桔色不溶物;反應液抽濾,濾餅採用丙酮進行反覆洗滌去除殘留溶劑及未反應的丁二酸酐,而後將濾餅溶於水並經冷凍乾燥得淡黃色海綿狀固體,即SA-PEG-FA;
採用上述同樣的合成方法,將前述步驟(2)中所製備的FA-PEG-NH2更換為1質量份的步驟(2)中所製備的TO-PEG-NH2,反應步驟及其他原料用量同上;不同在於,反應結束後,反應液經旋蒸濃縮後逐滴滴入冷乙醚中,析出紅色不溶物,過濾除去溶劑,濾餅用去離子水進行洗滌除去未反應的丁二酸酐,而後將濾液置於分子量為2000的透析袋中進行透析,而將透析袋中反應液後經冷凍乾燥得淺紅色海綿狀固體,即SA-PEG-TO。
(4)兼具靶向性和螢光特性的pH敏感殼聚糖載藥膠束(FA-PEG-OCCC-PEG-TO)的合成
稱取1質量份的步驟(3)中所製備的SA-PEG-FA和1質量份的步驟(3)中所製備的SA-PEG-TO,並將它們在無水二甲基亞碸(DMSO)中進行溶脹,而後以0.3-0.6質量份的DCC和0.2-0.4質量份的NHS為縮合劑,室溫下避光攪拌3-5h;而後向反應液中加入1-2質量份的步驟(1)中所製備的OCCC反應24h;反應結束後,向體系中加入蒸餾水,過濾除去不溶物,所得濾液置於分子量為3500的透析袋中進行透析,而後透析袋中的反應液經冷凍乾燥得橘紅色海綿狀產物,即兼具靶向性和螢光特性的pH敏感殼聚糖載藥膠束(FA-PEG-OCCC-PEG-TO)。
所述螢光染料噻唑橙(TO)的分子結構式如下所示:
本發明的有益效果是:
(1)本發明的殼聚糖載藥膠束具有良好的螢光性能,濃度為1mg/mL殼聚糖載藥膠束溶液,在480nm的激發波長下,其螢光強度值達到175a.u.。
(2)本發明的殼聚糖載藥膠束對抗癌藥5-氟尿嘧啶具有良好的載藥性能,在濃度為10mg/mL的殼聚糖膠束溶液對5-氟尿嘧啶的載藥效率可達42.76%。此外,本發明的殼聚糖載藥膠束的pH敏感範圍為4.0-4.5,與腫瘤癌細胞溶酶體pH值相近。載藥膠束在生理環境pH值(pH=7.4)條件下,其在8h內累積釋放藥量僅為2%,26h累積釋藥量不足8%,從而可減少殼聚糖載藥膠束在輸運抗癌藥5-氟尿嘧啶對正常組織細胞的毒副作用。同時,在腫瘤細胞溶酶體pH(pH=4.5)條件下,所製備殼聚糖載藥膠束表現出良好的pH敏感釋藥性能,5-氟尿嘧啶的釋放量在4h時達到95%。
附圖說明
圖1為本發明殼聚糖載藥膠束FA-PEG-(OCCC)-PEG-TO的合成路線圖。
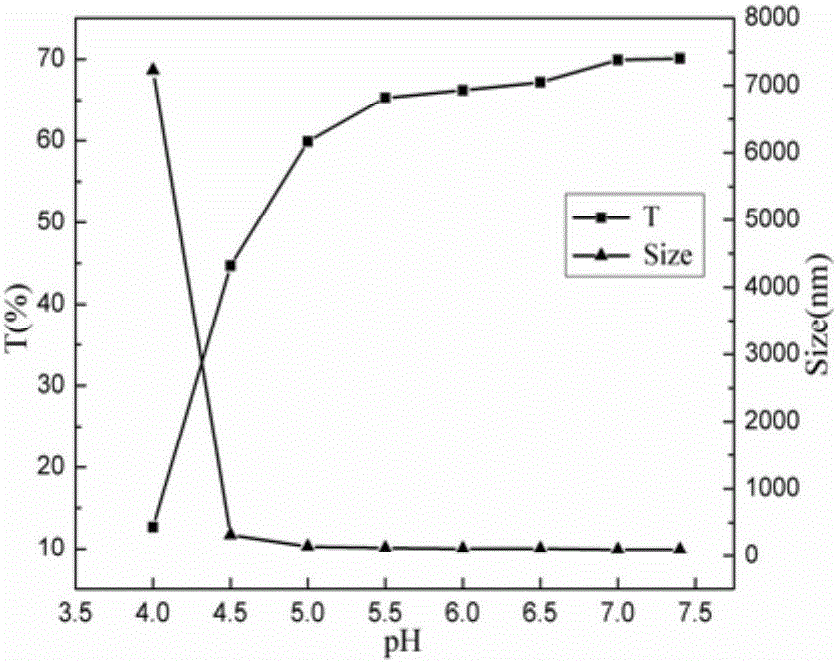
圖2為TO-PEG-(OCCC-2)-PEG-FA在不同pH緩衝溶液中的粒徑與透過率圖。
圖3為生理環境pH(pH=7.4)條件下TO-PEG-(OCCC-2)-PEG-FA膠束中5-氟尿嘧啶的緩釋效果圖。
圖4為在腫瘤細胞溶酶體pH值(pH=4.0)條件下TO-PEG-(OCCC-2)-PEG-FA膠束中5-氟尿嘧啶的釋放效果圖。
具體實施方式
下面結合附圖和具體實施方式對本發明作進一步詳細說明:
本發明的兼具靶向性和螢光的pH敏感殼聚糖載藥膠束的構建思想是,以具有pH敏感性的兩親性N-辛基-N』-(2-羧基環己甲醯基)殼聚糖(OCCC)為載藥膠束,以聚乙二醇為連接臂,分別在其兩端接枝葉酸和螢光染料噻唑橙,從而構建得到一種集靶向性、pH敏感性、隱形性和螢光示蹤於一體的多功能殼聚糖載藥膠束。在該殼聚糖載藥膠束的製備過程中,先以殼聚糖為主體,通過還原胺化反應在其上接枝疏水的辛基鏈,在以酸敏感β-羧酸醯胺鍵為連接臂接枝親水的羧基環己甲醯基,從而構建得到具有pH敏感特性的兩親性殼聚糖;然後,在端氨基聚乙二醇(H2N-PEG-NH2)分別通過縮合反應接枝葉酸(FA)和螢光染料噻唑橙(TO)製備得到FA-PEG-NH2和TO-PEG-NH2;再者,通過—反應在所製備的FA-PEG-NH2和TO-PEG-NH2的氨基端分別接枝丁二酸酐(SA)而製備得到FA-PEG-SA和TO-PEG-SA;最後,在縮合劑作用下,通過FA-PEG-SA和TO-PEG-SA中的酸酐與兩親性pH敏感殼聚糖上氨基之間的縮合反應,製備得到兼具靶向性和螢光特性的pH敏感殼聚糖載藥膠束(FA-PEG-OCCC-PEG-TO),具體合成路線圖如圖1所示,其具體製備方法主要包括以下幾個步驟:
(1)N-辛基-N』-(2-羧基環己甲醯基)殼聚糖的合成
稱取1質量份的殼聚糖(CTS),並將其置於無水甲醇(MeOH)中,在30℃和機械攪條件下進行溶脹;而後向反應液中緩慢滴加2.07-2.48質量份的辛醛,保溫反應5-36h後,分六個批次加入硼氫化鈉0.5-1質量份的NaBH4,室溫下繼續還原12-36h,還原反應完成後調節反應液pH值至6-8;反應液抽濾,並用甲醇和去離子水交替洗滌濾餅,而後濾餅經真空乾燥後得到淡黃色的N-辛基殼聚糖(OC);
稱取1質量份的前述N-辛基殼聚糖(OC)並將其置於反應瓶中,採用無水二甲基亞碸(DMSO)進行溶脹,在室溫條件下向反應瓶中滴加1-2質量份的六氫苯酐(C8H10O3),升溫至80℃反應12-36h;反應結束後,將反應液倒入去離子水中並置於冰水浴中,並調節反應液pH值至9-10;反應液進行抽濾,將濾液置於分子量為3500的透析袋中進行透析,而後將透析袋中反應液經冷凍乾燥即可得黃白色絮狀固體N-辛基-N』-(2-羧基環己甲醯基)殼聚糖(OCCC);
(2)聚乙二醇的葉酸接枝物(FA-PEG-NH2)及噻唑橙接枝物(TO-PEG-NH2)的合成
以二甲基亞碸為溶劑,稱取1質量份的葉酸(FA),並以1.38-1.72質量份的N-羥基琥珀醯亞胺(NHS)和0.35-0.70質量份的二環己基碳二亞胺(DCC)為縮合劑,在室溫下避光攪拌4h,而後向反應體系中加入3.45-6.90質量份的端氨基聚乙二醇(NH2-PEG-NH2),繼續攪拌反應8-12h。反應結束後向體系加入去離子水,抽濾除去不溶物,所得濾液離心後進行冷凍乾燥;冷凍乾燥所得固體產物在氯仿中溶解並進行抽濾,濾液用乙醚進行重結晶,過濾所得析出物再次溶於去離子水中,並再次經冷凍乾燥得橘黃色海綿狀產物,即聚乙二醇的葉酸接枝物(FA-PEG-NH2);
採用同樣的合成方法,將前述葉酸(FA)換成1.38質量份的螢光染料噻唑橙(TO),其它反應條件不變,可得紅色海綿狀固體,即聚乙二醇的噻唑橙接枝物(TO-PEG-NH2);
(3)聚乙二醇的丁二酸酐(SA)接枝物的合成
稱取1質量份的步驟(2)中所製備的FA-PEG-NH2並將其溶於無水二甲基亞碸(DMSO)中,加入0.2-0.3質量份的丁二酸酐(SA),室溫下攪拌反應,反應結束後向反應液加入0.75-1.5質量份的氯仿,並將反應液滴入冷乙醚中析出桔色不溶物;反應液抽濾,濾餅採用丙酮進行反覆洗滌去除殘留溶劑及未反應的丁二酸酐,而後將濾餅溶於水並經冷凍乾燥得淡黃色海綿狀固體,即SA-PEG-FA;
採用上述同樣的合成方法,將前述步驟(2)中所製備的FA-PEG-NH2更換為1質量份的步驟(2)中所製備的TO-PEG-NH2,反應步驟及其他原料用量同上;不同在於,反應結束後,反應液經旋蒸濃縮後逐滴滴入冷乙醚中,析出紅色不溶物,過濾除去溶劑,濾餅用去離子水進行洗滌除去未反應的丁二酸酐,而後將濾液置於分子量為2000的透析袋中進行透析,而將透析袋中反應液後經冷凍乾燥得淺紅色海綿狀固體,即SA-PEG-TO。
(4)兼具靶向性和螢光特性的pH敏感殼聚糖載藥膠束(FA-PEG-OCCC-PEG-TO)的合成
稱取1質量份的步驟(3)中所製備的SA-PEG-FA和1質量份的步驟(3)中所製備的SA-PEG-TO,並將它們在無水二甲基亞碸(DMSO)中進行溶脹,而後以0.3-0.6質量份的DCC和0.2-0.4質量份的NHS為縮合劑,室溫下避光攪拌3-5h;而後向反應液中加入1-2質量份的步驟(1)中所製備的OCCC反應24h;反應結束後,向體系中加入蒸餾水,過濾除去不溶物,所得濾液置於分子量為3500的透析袋中進行透析,而後透析袋中的反應液經冷凍乾燥得橘紅色海綿狀產物,即兼具靶向性和螢光特性的pH敏感殼聚糖載藥膠束(FA-PEG-OCCC-PEG-TO)。
實施例1
以製備OC過程中保溫時間為5h所製備的OC-1為殼聚糖為主體所製備的殼聚糖載藥膠束FA-PEG-(OCCC-1)-PEG-TO為例
稱取4g殼聚糖(CTS),並將其置於無水甲醇(MeOH)中,在30℃和機械攪條件下進行溶脹;而後向反應液中緩慢滴加10mL的辛醛,保溫反應5h後,分六個批次加入硼氫化鈉2g NaBH4,室溫下繼續還原12h,還原反應完成後調節反應液pH值至6-8;反應液抽濾,並用甲醇和去離子水交替洗滌濾餅,而後濾餅經真空乾燥後得到淡黃色的N-辛基殼聚糖(OC);稱取1g前述N-辛基殼聚糖(OC)並將其置於反應瓶中,採用無水二甲基亞碸(DMSO)進行溶脹,在室溫條件下向反應瓶中滴加1g六氫苯酐,升溫至80℃反應12h;反應結束後,將反應液倒入去離子水中並置於冰水浴中,並調節反應液pH值至9-10;反應液進行抽濾,將濾液置於分子量為3500的透析袋中進行透析,而後將透析袋中反應液經冷凍乾燥即可得黃白色絮狀固體N-辛基-N』-(2-羧基環己甲醯基)殼聚糖(OCCC);
以二甲基亞碸為溶劑,稱取0.58g葉酸(FA),並以0.8g的N-羥基琥珀醯亞胺(NHS)和0.2g二環己基碳二亞胺(DCC)為縮合劑,在室溫下避光攪拌4h,而後向反應體系中加入2g端氨基聚乙二醇(NH2-PEG-NH2),繼續攪拌反應8h。反應結束後向體系加入去離子水,抽濾除去不溶物,所得濾液離心後進行冷凍乾燥;冷凍乾燥所得固體產物在氯仿中溶解並進行抽濾,濾液用乙醚進行重結晶,過濾所得析出物再次溶於去離子水中,並再次經冷凍乾燥得橘黃色海綿狀產物,即聚乙二醇的葉酸接枝物(FA-PEG-NH2);採用同樣的合成方法,將前述葉酸(FA)換成0.8g螢光染料噻唑橙(TO),其它反應條件不變,可得紅色海綿狀固體,即聚乙二醇的噻唑橙接枝物(TO-PEG-NH2);
稱取2g所製備的FA-PEG-NH2並將其溶於無水二甲基亞碸(DMSO)中,加入0.4g丁二酸酐(SA),室溫下攪拌反應,反應結束後向反應液加入1mL氯仿,並將反應液滴入冷乙醚中析出桔色不溶物;反應液抽濾,濾餅採用丙酮進行反覆洗滌去除殘留溶劑及未反應的丁二酸酐,而後將濾餅溶於水並經冷凍乾燥得淡黃色海綿狀固體,即SA-PEG-FA;採用上述同樣的合成方法,將前述FA-PEG-NH2更換為2g所製備的TO-PEG-NH2,反應步驟及其他原料用量同上;不同在於,反應結束後,反應液經旋蒸濃縮後逐滴滴入冷乙醚中,析出紅色不溶物,過濾除去溶劑,濾餅用去離子水進行洗滌除去未反應的丁二酸酐,而後將濾液置於分子量為2000的透析袋中進行透析,而將透析袋中反應液後經冷凍乾燥得淺紅色海綿狀固體,即SA-PEG-TO。
稱取1.2g所製備的SA-PEG-FA和1.2g SA-PEG-TO,並將它們在無水二甲基亞碸(DMSO)中進行溶脹,而後以0.36g DCC和0.24g NHS為縮合劑,室溫下避光攪拌3h;而後向反應液中加入1.2g之前所製備的OCCC反應24h;反應結束後,向體系中加入蒸餾水,過濾除去不溶物,所得濾液置於分子量為3500的透析袋中進行透析,而後透析袋中的反應液經冷凍乾燥得橘紅色海綿狀產物,即兼具靶向性和螢光特性的pH敏感殼聚糖載藥膠束FA-PEG-(OCCC-1)-PEG-TO。
實施例2
以製備OC過程中保溫時間為8h所製備的OC-2為殼聚糖為主體所製備的殼聚糖載藥膠束FA-PEG-(OCCC-2)-PEG-TO為例
稱取4g殼聚糖(CTS),並將其置於無水甲醇(MeOH)中,在30℃和機械攪條件下進行溶脹;而後向反應液中緩慢滴加11.5mL的辛醛,保溫反應8h後,分六個批次加入硼氫化鈉2g NaBH4,室溫下繼續還原20h,還原反應完成後調節反應液pH值至6-8;反應液抽濾,並用甲醇和去離子水交替洗滌濾餅,而後濾餅經真空乾燥後得到淡黃色的N-辛基殼聚糖(OC);稱取1g前述N-辛基殼聚糖(OC)並將其置於反應瓶中,採用無水二甲基亞碸(DMSO)進行溶脹,在室溫條件下向反應瓶中滴加1.2g六氫苯酐,升溫至80℃反應20h;反應結束後,將反應液倒入去離子水中並置於冰水浴中,並調節反應液pH值至9-10;反應液進行抽濾,將濾液置於分子量為3500的透析袋中進行透析,而後將透析袋中反應液經冷凍乾燥即可得黃白色絮狀固體N-辛基-N』-(2-羧基環己甲醯基)殼聚糖(OCCC);
以二甲基亞碸為溶劑,稱取0.58g葉酸(FA),並以0.85gN-羥基琥珀醯亞胺(NHS)和0.36g二環己基碳二亞胺(DCC)為縮合劑,在室溫下避光攪拌4h,而後向反應體系中加入3g端氨基聚乙二醇(NH2-PEG-NH2),繼續攪拌反應10h。反應結束後向體系加入去離子水,抽濾除去不溶物,所得濾液離心後進行冷凍乾燥;冷凍乾燥所得固體產物在氯仿中溶解並進行抽濾,濾液用乙醚進行重結晶,過濾所得析出物再次溶於去離子水中,並再次經冷凍乾燥得橘黃色海綿狀產物,即聚乙二醇的葉酸接枝物(FA-PEG-NH2);採用同樣的合成方法,將前述葉酸(FA)換成0.8g螢光染料噻唑橙(TO),其它反應條件不變,可得紅色海綿狀固體,即聚乙二醇的噻唑橙接枝物(TO-PEG-NH2);
稱取2g所製備的FA-PEG-NH2並將其溶於無水二甲基亞碸(DMSO)中,加入0.5g丁二酸酐(SA),室溫下攪拌反應,反應結束後向反應液加入1.5mL氯仿,並將反應液滴入冷乙醚中析出桔色不溶物;反應液抽濾,濾餅採用丙酮進行反覆洗滌去除殘留溶劑及未反應的丁二酸酐,而後將濾餅溶於水並經冷凍乾燥得淡黃色海綿狀固體,即SA-PEG-FA;採用上述同樣的合成方法,將前述FA-PEG-NH2更換為2g所製備的TO-PEG-NH2,反應步驟及其他原料用量同上;不同在於,反應結束後,反應液經旋蒸濃縮後逐滴滴入冷乙醚中,析出紅色不溶物,過濾除去溶劑,濾餅用去離子水進行洗滌除去未反應的丁二酸酐,而後將濾液置於分子量為2000的透析袋中進行透析,而將透析袋中反應液後經冷凍乾燥得淺紅色海綿狀固體,即SA-PEG-TO。
稱取1.2g所製備的SA-PEG-FA和1.2g SA-PEG-TO,並將它們在無水二甲基亞碸(DMSO)中進行溶脹,而後以0.48g DCC和0.32g NHS為縮合劑,室溫下避光攪拌4h;而後向反應液中加入2g之前所製備的OCCC反應24h;反應結束後,向體系中加入蒸餾水,過濾除去不溶物,所得濾液置於分子量為3500的透析袋中進行透析,而後透析袋中的反應液經冷凍乾燥得橘紅色海綿狀產物,即兼具靶向性和螢光特性的pH敏感殼聚糖載藥膠束FA-PEG-(OCCC-2)-PEG-TO。
實施例3
以製備OC過程中保溫時間為36h所製備的OC-3為殼聚糖為主體所製備的殼聚糖載藥膠束FA-PEG-(OCCC-3)-PEG-TO為例
稱取4g殼聚糖(CTS),並將其置於無水甲醇(MeOH)中,在30℃和機械攪條件下進行溶脹;而後向反應液中緩慢滴加12mL的辛醛,保溫反應36h後,分六個批次加入硼氫化鈉4g NaBH4,室溫下繼續還原36h,還原反應完成後調節反應液pH值至6-8;反應液抽濾,並用甲醇和去離子水交替洗滌濾餅,而後濾餅經真空乾燥後得到淡黃色的N-辛基殼聚糖(OC);稱取1g前述N-辛基殼聚糖(OC)並將其置於反應瓶中,採用無水二甲基亞碸(DMSO)進行溶脹,在室溫條件下向反應瓶中滴加2g六氫苯酐,升溫至80℃反應36h;反應結束後,將反應液倒入去離子水中並置於冰水浴中,並調節反應液pH值至9-10;反應液進行抽濾,將濾液置於分子量為3500的透析袋中進行透析,而後將透析袋中反應液經冷凍乾燥即可得黃白色絮狀固體N-辛基-N』-(2-羧基環己甲醯基)殼聚糖(OCCC);
以二甲基亞碸為溶劑,稱取0.58g葉酸(FA),並以1g N-羥基琥珀醯亞胺(NHS)和0.4g二環己基碳二亞胺(DCC)為縮合劑,在室溫下避光攪拌4h,而後向反應體系中加入4g端氨基聚乙二醇(NH2-PEG-NH2),繼續攪拌反應12h。反應結束後向體系加入去離子水,抽濾除去不溶物,所得濾液離心後進行冷凍乾燥;冷凍乾燥所得固體產物在氯仿中溶解並進行抽濾,濾液用乙醚進行重結晶,過濾所得析出物再次溶於去離子水中,並再次經冷凍乾燥得橘黃色海綿狀產物,即聚乙二醇的葉酸接枝物(FA-PEG-NH2);採用同樣的合成方法,將前述葉酸(FA)換成0.8g螢光染料噻唑橙(TO),其它反應條件不變,可得紅色海綿狀固體,即聚乙二醇的噻唑橙接枝物(TO-PEG-NH2);
稱取2g所製備的FA-PEG-NH2並將其溶於無水二甲基亞碸(DMSO)中,加入0.6g丁二酸酐(SA),室溫下攪拌反應,反應結束後向反應液加入2mL氯仿,並將反應液滴入冷乙醚中析出桔色不溶物;反應液抽濾,濾餅採用丙酮進行反覆洗滌去除殘留溶劑及未反應的丁二酸酐,而後將濾餅溶於水並經冷凍乾燥得淡黃色海綿狀固體,即SA-PEG-FA;採用上述同樣的合成方法,將前述FA-PEG-NH2更換為2g所製備的TO-PEG-NH2,反應步驟及其他原料用量同上;不同在於,反應結束後,反應液經旋蒸濃縮後逐滴滴入冷乙醚中,析出紅色不溶物,過濾除去溶劑,濾餅用去離子水進行洗滌除去未反應的丁二酸酐,而後將濾液置於分子量為2000的透析袋中進行透析,而將透析袋中反應液後經冷凍乾燥得淺紅色海綿狀固體,即SA-PEG-TO。
稱取1.2g所製備的SA-PEG-FA和1.2g SA-PEG-TO,並將它們在無水二甲基亞碸(DMSO)中進行溶脹,而後以0.72g DCC和0.48g NHS為縮合劑,室溫下避光攪拌5h;而後向反應液中加入2.4g之前所製備的OCCC反應24h;反應結束後,向體系中加入蒸餾水,過濾除去不溶物,所得濾液置於分子量為3500的透析袋中進行透析,而後透析袋中的反應液經冷凍乾燥得橘紅色海綿狀產物,即兼具靶向性和螢光特性的pH敏感殼聚糖載藥膠束FA-PEG-(OCCC-3)-PEG-TO。
實施例4
以實施例2中所製備的殼聚糖載藥膠束FA-PEG-(OCCC-2)-PEG-TO為例測定其螢光性能和pH敏感特性。
將實施例2中所製備的殼聚糖載藥膠束FA-PEG-(OCCC-2)-PEG-TO配置成濃度為1mg/mL的溶液,取溶液10mL並將其超聲分散後採用螢光分光光度計測定其螢光譜,激發波長為480nm;測試結果發現,所製備殼聚糖載藥膠束的螢光發射波長為539nm,其螢光強度為175a.u.。
為測定殼聚糖載藥膠束FA-PEG-(OCCC-2)-PEG-TO的pH敏感特性,將其配製濃度為10mg/mL的溶液,取1ml溶液分別滴加至8mL pH=7.4,7.0,6.5,6.0,5.5,5.0,4.5,4.0的磷酸鹽緩衝溶液中,而後通過紫外-可見分光光度計測定溶液在500nm處的透過率,並用Nano Zeta Sizer電位粒徑儀測定空白膠束的粒徑,以溶液透過率和膠束粒徑大小來對膠束pH敏感特性進行測定,結果如圖2所示。可以發現,當緩衝溶液的pH值降低至4-4.5時,溶液透過率出現驟然降低的現象,下降至10%左右,且膠束粒徑也出現突然增大的現象,由500nm左右直接增加到4000nm,說明所製備的殼聚糖載藥膠束在溶液pH值為4-4.5時,其膠束結構遭到破壞,開始從水溶液中析出並凝聚成絮狀物。上述實驗結果說明所製備的殼聚糖載藥膠束FA-PEG-(OCCC-2)-PEG-TO的pH響應範圍為4-4.5。
實施實例5
以實施例2中所製備的殼聚糖載藥膠束FA-PEG-(OCCC-2)-PEG-TO為例測定其載藥和pH敏感釋藥特性。
採用pH=7.4的磷酸鹽緩衝溶液,分別配製濃度為1mg/mL、5mg/mL和10mg/mL的三組FA-PEG-(OCCC-2)-PEG-TO溶液各10mL,並分別向其中加入10mg 5-氟尿嘧啶,超聲分散5h後分別移入透析袋(Mw=3500)置於200mL磷酸鹽緩衝溶液裡室溫下透析40h除去游離5-氟尿嘧啶,每10h換透析液一次,並分別於20h,30h,40h取樣透析介質,通過紫外分光光度計測定溶液在265nm處的吸光度A,按回歸方程計算溶液中5-氟尿嘧啶濃度,直至不再透析出遊離5-氟尿嘧啶,取出透析袋內液體得到載藥膠束溶液。測定所得載藥膠束溶液在265nm處的吸光度A,計算載藥量。結果發現,1mg/mL、5mg/mL和10mg/mL的三組FA-PEG-(OCCC-2)-PEG-TO溶液對抗癌藥5-氟尿嘧啶的載藥效率依次為8.67%,17.56%和42.76%。
取4mL製得的濃度為10mg/mL的載藥膠束溶液兩份移入兩個透析袋(Mw=3500)分別置於20mL的pH=7.4和pH=4.5的磷酸鹽緩衝溶液中,恆溫37±0.5℃透析48h,並分別於2h,4h,8h,12h,16h,20h,26h,40h,48h抽取透析袋外液體3mL,並立即補充等溫等pH磷酸鹽緩衝液3mL。紫外分光光度計測定抽取溶液在265nm處的吸光度,根據線性回歸方程計算釋藥量,並對時間t作圖,測試結果如圖3和圖4所示。
從圖3中可以看出,所製備的殼聚糖載藥膠束在生理環境pH值(pH=7.4)條件下,其在8h內累積釋放藥量僅為2%,26h累積釋藥量不足8%;對比圖4可以看出,在腫瘤細胞溶酶體pH值(pH=4.5)條件下,所製備殼聚糖載藥膠束表現出良好的pH敏感釋藥性能,其累積藥物釋放量在1.5h已達到85%,4h的累積藥物釋放量最終達到95%,表現出良好pH敏感釋藥性能。
綜上所述,本發明的內容並不局限在上述的實施例中,相同領域內的有識之士可以在本發明的技術指導思想之內可以輕易提出其他的實施例,但這種實施例都包括在本發明的範圍之內。