一種製備含末端苯氰二聚體的方法與流程
2023-06-14 03:48:01 2
本發明涉及一種液晶化合物的準備方法,尤其涉及一種製備含末端苯氰二聚體的方法。
背景技術:
為了進一步提高液晶顯示器件的響應速度,近年來,開發出了基於液晶撓曲電效應的顯示模式。液晶的撓曲電效應具有非常快的響應速度,一般在10微秒~100微秒之間,比傳統的液晶顯示模式響應速度快1~2個數量級。如AppliedPhysicsLetters,2012,100,063501報導了基於液晶撓曲電效應的顯示模式,在該顯示器件中,液晶分子以「均勻臥式螺旋」(Uniformly LyingHelix,ULH)的方式排列,這種模式也稱之為ULH模式。採用ULH模式的液晶器件響應速度達到100微秒。隨後在ULH模式基礎上,又提出了「均勻豎立螺旋」(UniformlyStandingHelix,USH),與ULH模式相比,USH模式可以達到更佳的暗態,同時還能獲得寬的可視角範圍。傳統的液晶顯示模式要求液晶材料具有大的介電各向異性值(Δε),以降低顯示器的驅動電壓。而ULH/USH模式不同,為了防止液晶的螺旋結構在電場下解開,往往要求液晶材料具有儘可能小的介電各向異性值;同時為了降低驅動電壓,還需要液晶材料具有大的撓曲電係數。JournalofAppliedPhysics,1994,76,3762的論文研究結果表明,分子中包含兩個液晶基元的化合物具有大的撓曲電係數,可用於撓曲電效應的顯示模式。該類液晶也稱為二聚體化合物。此外,為了獲得低的介電各向異性值,分子一般應為對稱結構;同時,為了獲得較佳的撓曲電-光效應,二聚體化合物的中心橋鍵一般為奇數原子。
迄今為止,已經開發出多種二聚體化合物,並已在撓曲電液晶顯示模式中得到應用。Proceedings ofthe National Academy of Sciences of the United States of America,110(40),15931-15936,S15931/1-S15931/24,2013報導了二聚體的製備方法,但該方法步驟冗長(5步),收率低。鑑於二聚體化合物具有如此廣泛的應用領域,且應用前景廣闊,故仍需尋找一種步驟簡單、原料易得,高產率的二聚體化合物合成方法。
技術實現要素:
發明目的:本發明的目的是提供一種工藝設計合理,步驟簡單、原料易得,產率高可應用於撓曲電液晶顯示模式中的含末端苯氰二聚體(式M化合物)的製備方法。
技術方案:為解決上述問題,本發明提供如下技術方案:
本發明提供一種製備所述式M化合物的方法,
所述方法包括以下步驟:
1)將式1的化合物和有機溶劑在氮氣保護並溫控0℃-10℃下,分5-10次加入Lewis酸,攪拌1-30min後,升溫至30-50℃回流1-5小時後,降溫至10-20℃,
然後加入式2的化合物攪拌1-30min,升溫至30-50℃回流5-20h,
得到式3的化合物
2)將式3的化合物和強酸類溶劑混合,控溫20℃-30℃並在攪拌中滴加試劑三乙基矽烷,攪拌1-30min,升溫至40-60℃回流5-20小時,得到式4的化合物
3)將式4的化合物和式5的化合物混合,
在鹼性化合物、催化劑、水合肼、四氫呋喃和水的作用下反應,攪拌中氮氣置換3-8次後,升溫至70℃-95℃回流1-5小時,得到式M所示的化合物;
其中,n表示3、5、7或9。
在本發明的實施方案中,優選地所述有機溶劑為二氯甲烷或二氯乙烷。
在本發明的實施方案中,優選地所述Lewis酸為無水氯化鋁或無水氯化鐵。
在本發明的實施方案中,優選地所述強酸類溶劑為三氟乙酸或三氟甲磺酸。
在本發明的實施方案中,優選地所述鹼性化合物為碳酸鉀、碳酸鈉、氫氧化鉀或氫氧化鈉,所述催化劑為二(三苯基膦)二氯化鈀、四(三苯基膦)鈀或1,1'-雙二苯基膦二茂鐵二氯化鈀。
在本發明的實施方案中,優選地所述式1所示化合物與所述式2所示化合物、所述式3所示化合物、所述式4所示化合物、所述式5所示化合物的投料摩爾用量比為1:2:1:1:3。
在本發明的實施方案中,優選地所述式3所示化合物與強酸類有機溶劑的投料摩爾用量比為1:14.3。
在本發明的實施方案中,優選地所述式3所示化合物與試劑三乙基矽烷的投料摩爾用量比為1:4。
在本發明的實施方案中,優選地所述4所示化合物與所述鹼性化合物的投料摩爾用量比為1:4。
在本發明的實施方案中,優選地所述4所示化合物與所述催化劑、水合肼的投料摩爾用量比為1:0.02:0.045。
有益效果:本發明提供式M化合物的製備方法,整個工藝設計合理,可操作性強,原料便宜易得,合成路線簡單易行,產率高,適合規模化工業生產,可應用於撓曲電液晶顯示模式中,性能優越。
附圖說明
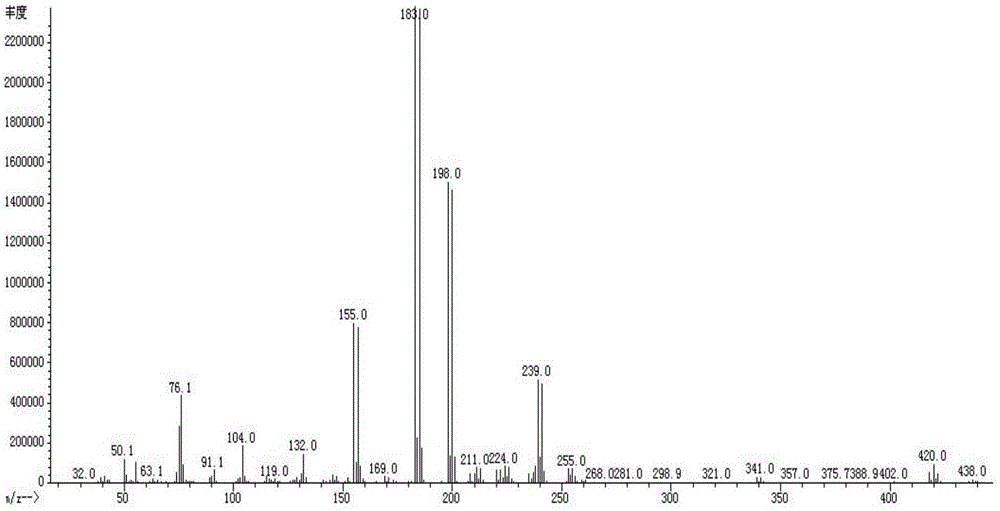
圖1是式3化合物的MS圖譜。
圖2是式4化合物的MS圖譜。
圖3是式M化合物的MS圖譜。
圖4是式M化合物的H譜圖。
具體實施方式
以下將結合具體實施方案來說明本發明。需要說明的是,下面的實施例為本發明的示例,僅用來說明本發明,而不用來限制本發明。在不偏離本發明主旨或範圍的情況下,可進行本發明構思內的其他組合和各種改良。
本發明所使用的試劑以及其他原料均為市售的商業原料。
實施例1
製備式M化合物的合成路線如下表示,
其具體工藝步驟如下:
1)化合物3的製備
於反應瓶中加入39g式1的化合物,300ml二氯甲烷,在氮氣保護並控溫10℃以下,分7~8次,加入58.7g無水氯化鋁。在溫度10℃以下攪拌30min後,升溫至40℃後回流1h,再降溫至20℃,10min內滴加62.4g式2的化合物,攪拌30min,再升溫至40℃後回流反應10h。
燒杯中倒入400ml冰水與20ml濃鹽酸,攪拌中倒入上述反應液,攪拌分層,分液,水相用100ml二氯甲烷萃取兩次,合併有機層,用無水硫酸鈉乾燥24h,蒸乾溶劑。墊50g 200~300目矽膠,用500ml乙酸乙酯淋洗,減壓蒸乾溶劑,得到80.6g淺黃色固體得化合物3,收率:92%。化合物3的質譜圖如圖1所示。分子量為435.97。
2)化合物4的製備
將40.5g式3的化合物、150g三氟乙酸加入反應瓶中,控溫30℃以下並在攪拌中滴加43.1g三乙基矽烷,攪拌20min,再升溫至50℃反應10h。
將上述反應液在50℃減壓濃縮至100g左右,加入400ml水,加入200ml石油醚分液,水相用200ml石油醚萃取兩次,合併有機層,蒸乾得到74g黃色液體。墊50g 200~300目矽膠,用500ml石油醚衝淋,減壓濃縮得到30.3g淺黃色液體,得化合物4,收率:80%。化合物4的質譜圖如圖2所示。分子量為408.01。
3)化合物M的製備
將18g式4化合物、19.4g式5化合物、24.3g碳酸鉀、0.62g二(三苯基膦)二氯化鈀、0.1g水合肼、100ml四氫呋喃和88ml水加至反應瓶中,攪拌中氮氣置換5次後,再加熱至外溫90℃回流反應3h。
分液,水相用50ml乙酸乙酯萃取兩次,合併有機相,無水硫酸鈉乾燥,減壓濃縮得到29g黃褐色固體。然後墊50g 200~300目矽膠,用2L二氯甲烷:石油醚=1:1衝柱,減壓濃縮溶劑得到21g黃色固體。然後用二氯甲烷:石油醚=1:2重結晶3次,得到白色固體,為式M的化合物,收率:85%。化合物M的質譜圖如圖3所示。分子量為454.24。
化合物M的氫譜圖如圖4所示:1H-NMR(CDCl3)δ:7.671~7.748(m,8H);7.515~7.542(d,4H);7.281~7.320(m,4H);2.655~2.706(m,4H);1.585~1.673(m,4H);1.399(s,6H)。
檢測化合物M的液晶性能如下:Cp:54.47℃,Δn:0.256。
以上製備工藝設計合理,可操作性強,得率在80%以上,適合規模化工業生產,並且性能檢測結果表明,本發明製備得到的M化合物性能優異,可應用於撓曲電液晶顯示模式中。