一種改性雙馬來醯亞胺樹脂及其製備方法與流程
2023-06-06 04:46:51 4
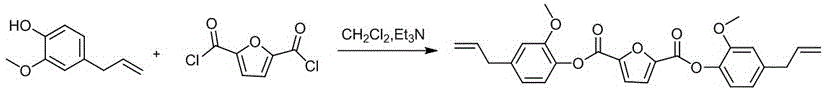
本發明涉及一種改性雙馬來醯亞胺樹脂及其製備方法,具體涉及一種利用綠色可再生生物質資源合成的一種基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯改性雙馬來醯亞胺樹脂,屬於化工與高分子材料技術領域。
背景技術:
在過去的幾十年中,石油和煤是生產燃料、化學藥品以及高分子材料等的重要原料。然而,石油和煤資源的不可再生性及人類可持續發展的緊迫性都要求開發出一種新型可再生的材料,而生物質材料恰好滿足人類的迫切需求。
生物質具有可再生、分布廣且年產量巨大的特點,但是迄今人們對其的利用效率非常低。如何高效地將可再生的生物質資源轉化為可利用的高分子材料,引發了整個世界的濃厚興趣並得到了高度關注。自然界廣泛存在的生物質材料多為脂肪族化合物,熱學性能較差,因此,丁香酚和2,5-呋喃二甲酸以其芳香結構而具有的優異熱穩定性脫穎而出。丁香酚約佔丁香油80%的成分,是一種可再生、低毒、相對低成本(大約每公斤5美元)的生物質材料。2,5-呋喃二甲酸被U.S. Department of Energy列入排名前十的綠色化學物質,已經用於合成尼龍、聚酯和聚氨酯等。然而,在科技進步和經濟發展的要求下,高性能樹脂以其優異的性能可應用於高端領域,近年來得到了廣泛深入的研究,但是2,5-呋喃二甲酸和丁香酚在高性能樹脂方面應用較少。
作為高性能熱固性樹脂的典型代表,雙馬來醯亞胺樹脂(BMI)的固化物具有優異的綜合性能(包括突出的抗熱氧化性和耐熱性、優異的力學性能、良好的耐溼熱及優異的介電性能等),因而在航空、電子信息、電氣絕緣等高端領域佔有重要地位。但未改性的BMI存在熔點高、溶解性差、成型溫度高,固化物交聯密度太大、脆性大等缺點,不能很好地滿足加工和使用的要求。迄今為止,烯丙基苯基化合物改性BMI是BMI改性中工藝最成熟、效果最好的一種,是國內外BMI改性研究的熱點,目前對其研究大多針對是4,4』-雙馬來醯亞胺基二苯甲烷(BDM)進行的。2,2』-二烯丙基雙酚A(DBA)是最常用的BDM的烯丙烯基苯基化合物改性劑。然而它是通過雙酚A二烯丙基醚(BBE)在高溫下發生克萊森重排形成的,而BBE是通過以主要來自石油和煤資源的雙酚A和來自石油資源的氯丙烯或溴丙烯反應合成的。一方面,雙酚A來源於不可再生的石油和煤資源,在BDM改性中用量很大,這使它對石油和煤資源造成了很大依賴,不符合當今人類社會綠色可持續發展理念;另一方面,隨著生活水平提高,生產生活中人們對於健康和安全的要求日益增長,而雙酚A的類雌激素結構無形中會對健康產生極大的威脅,這使得全球使用非雙酚A基產品的呼聲日益高漲。因此,開發一種環保綠色低毒的基於生物質的BMI改性劑在資源保護和人類健康方面都大有裨益。
最近,已有少量文獻報導基於生物質的烯丙烯基苯基化合物用於改性BMI,然而它們中絕大多數並非使用全生物質原料合成。5,5』-二丁香酚(BEG)是迄今報導過的唯一一個用於BMI改性的基於全生物質的烯丙基苯基化合物的原料(參見文獻:Mitsuhiro Shibata, Naozumi Tetramoto, Ayumi Imada, Makiyo Neda, Shimon Sugimoto. Reactive & Functional Polymers, 2013, 73, 1086-1095),但是所合成BEG的轉化率只有60%。此外,其改性BDM樹脂不能兼具高彎曲模量、高玻璃化轉變溫度(Tg)和高起始熱分解溫度(Tdi)。當BEG與BDM按摩爾比為0.5:1時,改性樹脂的Tg為285.8℃,Tdi為423.1℃,但是彎曲模量只有2.7 GPa;而當BEG與BDM摩爾比為1:1時,改性樹脂的Tg只有201.2℃,Tdi為419.0℃,彎曲模量小於1 GPa。熱性能和彎曲模量都低於基於石油和煤資源的DBA改性BMI樹脂。這是因為BEG是通過兩分子的丁香酚直接連接在一起得到的,分子結構呈現聯苯結構,位阻大,使得BDM與中間體雙鍵的Diels-Alder加成反應更難發生,致使固化物交聯密度降低,從而降低了熱性能和彎曲模量。
因此,製備一種基於全生物質的烯丙烯基苯基化合物,使改性BMI樹脂具有良好的熱性能和高彎曲模量,是一個具有重大應用價值的課題。
技術實現要素:
本發明針對現有技術存在的不足,提供一種同時具有良好的熱性能和高彎曲模量的改性雙馬來醯亞胺樹脂及其製備方法。
為達到上述發明目的,本發明所採用的技術方案是提供一種改性雙馬來醯亞胺樹脂的製備方法,包含如下步驟:
(1)按摩爾計,將100份 2,5呋喃二甲酸、150~250份二氯亞碸和催化量的N,N-二甲基甲醯胺混合,在溫度為70~80℃的條件下攪拌反應3~5h,自然冷卻至室溫,真空旋蒸去除二氯亞碸,乾燥後得到2,5呋喃二甲醯氯;
(2)按摩爾計,將190~210份丁香酚和240~300份叔胺溶解於3120~7800份的二氯甲烷中,得到丁香酚溶液A;在溫度為-5~0℃,攪拌條件下,將100份2,5呋喃二甲醯氯溶解於3120~7800份二氯甲烷中,再將得到的溶液滴加到丁香酚溶液A中,滴加完畢後,將反應液緩慢升溫至20~30℃,繼續反應2~4h,真空旋蒸去除二氯甲烷,經洗滌、乾燥,得到基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯;
(3)在溫度為20~30℃的條件下,按摩爾計,將1份雙馬來醯亞胺和0.55~1.20份二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯混合,在溫度為130~145℃的條件下,攪拌成透明液體,再經固化與後處理,即得到一種改性雙馬來醯亞胺樹脂。
本發明所述的叔胺為三乙胺、N-乙基二異丙胺、吡啶中的一種,或它們的任意組合。
所述的雙馬來醯亞胺為N, N』-4,4』-二苯醚雙馬來醯亞胺、N, N』-4,4』-二苯醚雙馬來醯亞胺的一種,或它們的任意組合。
本發明技術方案還包括按上述製備方法得到的一種改性雙馬來醯亞胺樹脂。
與現有技術提供的改性雙馬來醯亞胺樹脂不同,本發明所製備的改性雙馬來醯亞胺樹脂具有優良的熱性能和剛性,其原理是:本發明提供的全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯具有獨特的結構;一方面,丁香酚和2,5-呋喃二甲酸的芳香結構賦予所合成的全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯具有優異的熱穩定性,從而滿足高耐熱雙馬來醯亞胺樹脂的改性要求;另一方面,二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的烯丙基在化學結構的對位,位阻小,有利於 BDM與中間體雙鍵的Diels-Alder加成反應的發生,使得改性雙馬來醯亞胺樹脂具有更好的反應性,固化物具有高交聯密度,因此,製備的改性雙馬來醯亞胺樹脂具有優良的熱性能和剛性。
與現有技術相比,本發明取得的有益效果是:
1、本發明以生物質可再生資源丁香酚和2,5-呋喃二甲酸為原料,所製備的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的生物質含量高達100%,並且合成反應轉化率超過90%;用於對雙馬來醯亞胺樹脂進行改性,具有更好的反應性,固化物具有高交聯密度,因此,得到的改性雙馬來醯亞胺樹脂具有優良的熱性能和剛性。
2、與傳統石油基二烯丙基雙酚A的合成步驟相比,本發明提供的基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的合成過程無高溫重排過程,工藝簡單、節能。
3、與現有技術不同,本發明提供的改性雙馬來醯亞胺樹脂為非雙酚A基材料,因此,沒有雙酚A存在的降低生育能力、罹患疾病和癌症的風險。
4、本發明提供的一種改性雙馬來醯亞胺樹脂的製備方法具有工藝簡單,過程可控性好,易於工業化生產的特點。
附圖說明
圖1是本發明製備2,5呋喃二甲醯氯的合成反應式。
圖2是本發明製備基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的合成反應式。
圖3是本發明實施例1提供的基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的核磁共振氫譜。
圖4是本發明實施例1提供的基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的核磁共振碳譜。
圖5是本發明實施例1提供的基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的高分辨質譜圖。
圖6是本發明實施例3製備的改性雙馬來醯亞胺樹脂預聚體與比較例1製備的二烯丙基雙酚A改性雙馬來醯亞胺樹脂預聚體在氮氣氛圍下的差示掃描量熱曲線的對比圖。
圖7是本發明實施例3製備的改性雙馬來醯亞胺樹脂與比較例1製備的二烯丙基雙酚A改性雙馬來醯亞胺樹脂在氮氣氛圍下的熱失重曲線的對比圖。
圖8是本發明實施例3製備的改性雙馬來醯亞胺樹脂與比較例1製備的二烯丙基雙酚A改性雙馬來醯亞胺樹脂的彎曲模量的對比圖。
具體實施方式
下面結合附圖和實施例對本發明技術方案作進一步的描述。
實施例1
1)2,5呋喃二甲醯氯的製備
參見附圖1,它是本實施例中製備2,5呋喃二甲醯氯的合成反應式;具體的反應條件如下:
將31.20g 2,5呋喃二甲酸、35.69g二氯亞碸和N,N-二甲基甲醯胺(DMF,催化劑,0.05mL)混合,在溫度為80℃的條件下攪拌反應3h,自然冷卻至室溫,真空旋蒸掉二氯亞碸,乾燥後,得到2,5呋喃二甲醯氯,產率為99.5%。
2)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的製備
參見附圖2,它是本實施例中製備基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的合成反應式;具體的反應條件如下:
將31.20g丁香酚和24.29g三乙胺作為鹼溶解在200mL二氯甲烷中攪拌,在-5~0℃下滴入2,5呋喃二甲醯氯(19.30g)的二氯甲烷(200mL)溶液,滴加完畢後反應液緩慢升至20℃,繼續反應2h;反應結束後,真空旋蒸掉二氯甲烷,用去離子水洗滌,乾燥得到基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯,產率為91.2%。
在本實施例中得到的基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的核磁共振氫譜、核磁共振碳譜和高分辨質譜分別參見附圖3、4和5。
3)改性雙馬來醯亞胺樹脂的製備
在20℃下,將50.0g(139.5 mmol)N, N』-4,4』-二苯甲烷雙馬來醯亞胺和35.5g(76.73 mmol)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯混合,在溫度為130℃的恆溫條件下預聚反應30min;將所得到的預聚體倒入預熱好的模具中,在145℃的真空烘箱中抽氣30min, 再以150℃/2h+180℃/2h+200℃/2h和220℃/8h工藝分別進行固化和後處理,自然冷卻後脫模,即得固化的改性雙馬來醯亞胺樹脂。
由圖1反應式(合成流程示意圖)可見,本發明實施例1提供的中間體2,5呋喃二甲醯氯的合成,為羧酸醯氯化反應。由圖2反應式(合成流程示意圖)可見,本發明實施例1中基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的合成反應為酯化反應。
參見附圖3,它是本發明實施例1中基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的核磁共振氫譜,由圖可知,約5.98ppm和5.04~5.20ppm處代表烯丙基雙鍵上的H,約3.40ppm處代表烯丙基上與雙鍵相鄰的亞甲基上的H,約3.82ppm處代表甲氧基上的H,約7.43ppm處代表呋喃環上的H,其他峰與生物質烯丙基化合物的H質子位移相符。
參見附圖4,它是本發明實施例1中基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的核磁共振碳譜,由圖可知,約139.79ppm和116.46ppm峰分別代表基於全生物質的烯丙基化合物的烯丙基上的C。
參見附圖5,它是本發明實施例1製備的基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的高分辨質譜圖,其理論分子量[M]為448.1522,理論值[M+Na+]為471.1414,實驗值為471.1402,實驗值與理論值相符。
綜合以上附圖可知,本實施例1合成了預期物質基於全生物質的烯丙基化合物二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯。
實施例2
1)2,5呋喃二甲醯氯的製備
將31.20g 2,5呋喃二甲酸、35.69g二氯亞碸和N,N-二甲基甲醯胺(DMF,催化劑,0.05mL)混合,在溫度為80℃的條件下攪拌反應3h,自然冷卻至室溫,真空旋蒸掉二氯亞碸,乾燥後,得到2,5呋喃二甲醯氯,產率為99.5%。
2)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的製備
將32.84g丁香酚和27.33g三乙胺作為鹼溶解在300mL二氯甲烷中攪拌,在-2.5±1℃下滴入2,5呋喃二甲醯氯(19.30g)的二氯甲烷(300mL)溶液,滴加完畢後反應液緩慢升至20℃,繼續反應3h;反應結束後,真空旋蒸掉二氯甲烷,用去離子水洗滌,乾燥得到基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯,產率為91.2%。
3)改性雙馬來醯亞胺樹脂的製備
在25℃下,將50.0g(139.5 mmol) N, N』-4,4』-二苯甲烷雙馬來醯亞胺和44.0g(97.65 mmol)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯混合,在溫度為140℃的恆溫條件下預聚反應30min;將所得到的預聚體倒入預熱好的模具中,在145℃的真空烘箱中抽氣30min, 再以150℃/2h+180℃/2h+200℃/2h和250℃/5h工藝分別進行固化和後處理,自然冷卻後脫模,即得固化的改性雙馬來醯亞胺樹脂。
實施例3
1)2,5呋喃二甲醯氯的製備
將31.20g 2,5呋喃二甲酸、35.69g二氯亞碸和N,N-二甲基甲醯胺(DMF,催化劑,0.05mL)混合,在溫度為70oC的條件下攪拌反應3h,自然冷卻至室溫,真空旋蒸掉二氯亞碸,乾燥後,得到2,5呋喃二甲醯氯,產率為99.6%。
2)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的製備
將34.48g丁香酚和30.36g三乙胺作為鹼溶解在500mL二氯甲烷中攪拌,在-1±1oC下滴入2,5呋喃二甲醯氯(19.30g)的二氯甲烷(500mL)溶液,滴加完畢後反應液緩慢升至20℃,繼續反應4h;反應結束後,真空旋蒸掉二氯甲烷,用去離子水洗滌,乾燥得到基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯,產率為90.6%。
3)改性雙馬來醯亞胺樹脂的製備
在30℃下,將50.0g(139.5 mmol) N, N』-4,4』-二苯甲烷雙馬來醯亞胺和54.0g(120.0 mmol)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯混合,在溫度為145℃的恆溫條件下預聚反應30min;將所得到的預聚體倒入預熱好的模具中,在145℃的真空烘箱中抽氣30min, 再以150℃/2h+180℃/2h+200℃/2h+220℃/2h和240℃/4h工藝分別進行固化和後處理,自然冷卻後脫模,即得固化的改性雙馬來醯亞胺樹脂。其預聚物的差示掃描量熱曲線、改性雙馬來醯亞胺樹脂熱失重曲線圖和彎曲強度圖分別參見圖6、7和8。
比較例1,二烯丙基雙酚A改性雙馬來醯亞胺樹脂的製備:
在20℃下,將50.0g(139.5 mmol) N, N』-4,4』-二苯甲烷雙馬來醯亞胺、37.0g(120.0 mmol) 2, 2』-二烯丙基雙酚A混合,在溫度為145℃的恆溫條件下預聚反應30min,將所得到的預聚體倒入預熱好的模具中,在145℃的真空烘箱中抽氣30min, 再以150℃/2h+180℃/2h+200℃/2h+220℃/2h和240℃/4h工藝分別進行固化和後處理,自然冷卻後脫模,即得固化的二烯丙基雙酚A改性雙馬來醯亞胺樹脂。其預聚物的差示掃描量熱曲線、改性雙馬來醯亞胺樹脂熱失重曲線圖和彎曲模量圖分別參見圖6、7和8。
參見附圖6,為本發明實施例3製備的改性雙馬來醯亞胺樹脂預聚體與比較例1製備的二烯丙基雙酚A改性雙馬來醯亞胺樹脂預聚體在氮氣氛圍下的差示掃描量熱曲線的對比圖。由圖可知,實施例3製備的改性雙馬來醯亞胺樹脂預聚體的最大反應放熱峰為230.1℃,低於比較例1製備的二烯丙基雙酚A改性雙馬來醯亞胺樹脂預聚體的250.2℃;同時通過Kissinger方程計算,得到實施例3製備的改性雙馬來醯亞胺樹脂預聚體的反應活化能為67.5 kJ/mol,低於比較例1製備的二烯丙基雙酚A改性雙馬來醯亞胺樹脂預聚體的72.5 kJ/mol,表明實施例3製備的改性雙馬來醯亞胺樹脂的反應性更好。這是因為二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的烯丙基在化學結構的對位,位阻更小,所以反應性更好。
參見附圖7,為本發明實施例3製備的改性雙馬來醯亞胺樹脂與比較例1製備的二烯丙基雙酚A改性雙馬來醯亞胺樹脂在氮氣氛圍下的熱失重曲線的對比圖。一般用起始熱分解溫度(Tdi)表徵材料的熱穩定性。由圖可知,實施例3製備的改性雙馬來醯亞胺樹脂的Tdi為457.5℃,高於二烯丙基雙酚A改性雙馬來醯亞胺樹脂的431.5℃,表明改性雙馬來醯亞胺樹脂有更好的熱穩定性。比較例1製備的二烯丙基雙酚A改性雙馬來醯亞胺樹脂在800℃的殘炭量為25.1%,而實施例3製備的改性雙馬來醯亞胺樹脂在800℃的殘炭量高達為42.1%,這有利於獲得高阻燃。
參見附圖8,為本發明實施例3製備的改性雙馬來醯亞胺樹脂與比較例1製備的二烯丙基雙酚A改性雙馬來醯亞胺樹脂的彎曲模量的對比圖,由圖可知,實施例3製備的改性雙馬來醯亞胺樹脂的彎曲模量為4.17GPa,而比較例1製備的二烯丙基雙酚A改性雙馬來醯亞胺樹脂的彎曲模量為4.02GPa。這是因為實施例3製備的全生物質烯丙基化合物含有呋喃基團,有利於賦予改性樹脂優良的剛性。
實施例4
1)2,5呋喃二甲醯氯的製備
將31.20g 2,5呋喃二甲酸、35.69g二氯亞碸和N,N-二甲基甲醯胺(DMF,催化劑,0.05mL)混合,在溫度為70℃的條件下攪拌反應3h,自然冷卻至室溫,真空旋蒸掉二氯亞碸,乾燥後,得到2,5呋喃二甲醯氯,產率為99.6%。
2)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的製備
將31.20g丁香酚和31.03g N-乙基二異丙胺為鹼溶解在200mL二氯甲烷中攪拌,在-4±1℃下滴入2,5呋喃二甲醯氯(19.30g)的二氯甲烷(200mL)溶液,滴加完畢後反應液緩慢升至20℃,繼續反應2h;反應結束後,真空旋蒸掉二氯甲烷,用去離子水洗滌,乾燥得到基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯,產率為91.0%。
3)改性雙馬來醯亞胺樹脂的製備
在20℃下,將50.0g(139.5 mmol) N, N』-4,4』-二苯甲烷雙馬來醯亞胺和62.5g(139.5 mmol)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯混合,在溫度為140 oC的恆溫條件下預聚反應30min;將所得到的預聚體倒入預熱好的模具中,在145℃的真空烘箱中抽氣30min, 再以150℃/2h+180℃/2h+200℃/2h和220℃/8h工藝分別進行固化和後處理,自然冷卻後脫模,即得固化的改性雙馬來醯亞胺樹脂。
實施例5
1)2,5呋喃二甲醯氯的製備
將31.20g 2,5呋喃二甲酸、35.69g二氯亞碸和N,N-二甲基甲醯胺(DMF,催化劑,0.05mL)混合,在溫度為70oC的條件下攪拌反應3h,自然冷卻至室溫,真空旋蒸掉二氯亞碸,乾燥後,得到2,5呋喃二甲醯氯,產率為99.6%。
2)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的製備
將34.48g丁香酚和23.73g吡啶作為鹼溶解在500mL二氯甲烷中攪拌,在-1±1℃下滴入2,5呋喃二甲醯氯(19.30g)的二氯甲烷(500mL)溶液,滴加完畢後反應液緩慢升至20oC,繼續反應4h;反應結束後,真空旋蒸掉二氯甲烷,用去離子水洗滌,乾燥得到基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯,產率為90.8%。
3)改性雙馬來醯亞胺樹脂的製備
在30℃下,將50.0g(139.5 mmol) N, N』-4,4』-二苯甲烷雙馬來醯亞胺和75.0g(167.4 mmol)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯混合,在溫度為145℃的恆溫條件下預聚反應30min;將所得到的預聚體倒入預熱好的模具中,在145℃的真空烘箱中抽氣30min, 再以150℃/2h+180℃/2h+200℃/2h和250℃/5h工藝分別進行固化和後處理,自然冷卻後脫模,即得固化的改性雙馬來醯亞胺樹脂。
實施例6
1)2,5呋喃二甲醯氯的製備
將31.20g 2,5呋喃二甲酸、47.59g二氯亞碸和N,N-二甲基甲醯胺(DMF,催化劑,0.05mL)混合,在溫度為75℃的條件下攪拌反應4h,自然冷卻至室溫,真空旋蒸掉二氯亞碸,乾燥後,得到2,5呋喃二甲醯氯,產率為99.8%。
2)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的製備
將31.20g丁香酚和24.29g三乙胺作為鹼溶解在200mL二氯甲烷中攪拌,在-5~0℃下滴入2,5呋喃二甲醯氯(19.30g)的二氯甲烷(200mL)溶液,滴加完畢後反應液緩慢升至25℃,繼續反應2h;反應結束後,真空旋蒸掉二氯甲烷,用去離子水洗滌,乾燥得到基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯,產率為91.2%。
3)改性雙馬來醯亞胺樹脂的製備
在20℃下,將50.0g(139.5 mmol) N, N』-4,4』-二苯醚雙馬來醯亞胺和35.5g(76.73 mmol)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯混合,在溫度為130℃的恆溫條件下預聚反應30min;將所得到的預聚體倒入預熱好的模具中,在145℃的真空烘箱中抽氣30min, 再以150℃/2h+180℃/2h+200℃/2h和220℃/8h工藝分別進行固化和後處理,自然冷卻後脫模,即得固化的改性雙馬來醯亞胺樹脂。
實施例7
1)2,5呋喃二甲醯氯的製備
將31.20g 2,5呋喃二甲酸、47.59g二氯亞碸和N,N-二甲基甲醯胺(DMF,催化劑,0.05mL)混合,在溫度為75℃的條件下攪拌反應4h,自然冷卻至室溫,真空旋蒸掉二氯亞碸,乾燥後,得到2,5呋喃二甲醯氯,產率為99.8%。
2)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的製備
將32.84g丁香酚和27.33g三乙胺作為鹼溶解在300mL二氯甲烷中攪拌,在-2.5±1℃下滴入2,5呋喃二甲醯氯(19.30g)的二氯甲烷(300mL)溶液,滴加完畢後反應液緩慢升至25℃,繼續反應3h;反應結束後,真空旋蒸掉二氯甲烷,用去離子水洗滌,乾燥得到基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯,產率為91.8%。
3)改性雙馬來醯亞胺樹脂的製備
在25℃下,將50.0g(139.5 mmol) N, N』-4,4』-二苯醚雙馬來醯亞胺和44.0g(97.76 mmol)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯混合,在溫度為140 ℃的恆溫條件下預聚反應30min;將所得到的預聚體倒入預熱好的模具中,在145℃的真空烘箱中抽氣30min, 再以150℃/2h+180℃/2h+200℃/2h和250℃/5h工藝分別進行固化和後處理,自然冷卻後脫模,即得固化的改性雙馬來醯亞胺樹脂。
實施例8
1)2,5呋喃二甲醯氯的製備
將31.20g 2,5呋喃二甲酸、47.59g二氯亞碸和N,N-二甲基甲醯胺(DMF,催化劑,0.05mL)混合,在溫度為75℃的條件下攪拌反應4h,自然冷卻至室溫,真空旋蒸掉二氯亞碸,乾燥後,得到2,5呋喃二甲醯氯,產率為99.8%。
2)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的製備
將34.48g丁香酚和30.36g三乙胺作為鹼溶解在500mL二氯甲烷中攪拌,在-1±1℃下滴入2,5呋喃二甲醯氯(19.30g)的二氯甲烷(500mL)溶液,滴加完畢後反應液緩慢升至25℃,繼續反應4h;反應結束後,真空旋蒸掉二氯甲烷,用去離子水洗滌,乾燥得到基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯,產率為90.6%。
3)改性雙馬來醯亞胺樹脂的製備
在30℃下,將50.0g(139.5 mmol) N, N』-4,4』-二苯醚雙馬來醯亞胺和54.0g(120.0 mmol)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯混合,在溫度為145℃的恆溫條件下預聚反應30min;將所得到的預聚體倒入預熱好的模具中,在145℃的真空烘箱中抽氣30min, 再以150℃/2h+180℃/2h+200℃/2h+220℃/2h和240℃/4h工藝分別進行固化和後處理,自然冷卻後脫模,即得固化的改性雙馬來醯亞胺樹脂。
實施例9
1)2,5呋喃二甲醯氯的製備
將31.20g 2,5呋喃二甲酸、47.59g二氯亞碸和N,N-二甲基甲醯胺(DMF,催化劑,0.05mL)混合,在溫度為75℃的條件下攪拌反應4h,自然冷卻至室溫,真空旋蒸掉二氯亞碸,乾燥後,得到2,5呋喃二甲醯氯,產率為99.8%。
2)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的製備
將31.20g丁香酚和31.03g N-乙基二異丙胺為鹼溶解在200mL二氯甲烷中攪拌,在-4±1℃下滴入2,5呋喃二甲醯氯(19.30g)的二氯甲烷(200mL)溶液,滴加完畢後反應液緩慢升至25oC,繼續反應2h;反應結束後,真空旋蒸掉二氯甲烷,用去離子水洗滌,乾燥得到基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯,產率為91.0%。
3)改性雙馬來醯亞胺樹脂的製備
在20℃下,將50.0g(139.5 mmol)N, N』-4,4』-二苯醚雙馬來醯亞胺和62.5g(139.5 mmol)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯混合,在溫度為140℃的恆溫條件下預聚反應30min;將所得到的預聚體倒入預熱好的模具中,在145℃的真空烘箱中抽氣30min, 再以150℃/2h+180℃/2h+200℃/2h和220℃/8h工藝分別進行固化和後處理,自然冷卻後脫模,即得固化的改性雙馬來醯亞胺樹脂。
實施例10
1)2,5呋喃二甲醯氯的製備
將31.20g 2,5呋喃二甲酸、47.59g二氯亞碸和N,N-二甲基甲醯胺(DMF,催化劑,0.05mL)混合,在溫度為75℃的條件下攪拌反應4h,自然冷卻至室溫,真空旋蒸掉二氯亞碸,乾燥後,得到2,5呋喃二甲醯氯,產率為99.8%。
2)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的製備
將34.48g丁香酚和23.73g吡啶作為鹼溶解在500mL二氯甲烷中攪拌,在-1±1℃下滴入2,5呋喃二甲醯氯(19.30g)的二氯甲烷(500mL)溶液,滴加完畢後反應液緩慢升至25℃,繼續反應4h;反應結束後,真空旋蒸掉二氯甲烷,用去離子水洗滌,乾燥得到基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯,產率為90.8%。
3)改性雙馬來醯亞胺樹脂的製備
在30℃下,將50.0g(139.5 mmol) N, N』-4,4』-二苯醚雙馬來醯亞胺和75.0g(167.4 mmol)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯混合,在溫度為145℃的恆溫條件下預聚反應30min;將所得到的預聚體倒入預熱好的模具中,在145℃的真空烘箱中抽氣30min, 再以150℃/2h+180℃/2h+200℃/2h和250℃/5h工藝分別進行固化和後處理,自然冷卻後脫模,即得固化的改性雙馬來醯亞胺樹脂。
實施例11
1)2,5呋喃二甲醯氯的製備
將31.20g 2,5呋喃二甲酸、59.48g二氯亞碸和N,N-二甲基甲醯胺(DMF,催化劑,0.05mL)混合,在溫度為80℃的條件下攪拌反應5h,自然冷卻至室溫,真空旋蒸掉二氯亞碸,乾燥後得到2,5呋喃二甲醯氯,產率為99.6%。
2)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的製備
將31.20g丁香酚和24.29g三乙胺作為鹼溶解在200mL二氯甲烷中攪拌,在-5~0℃下滴入2,5呋喃二甲醯氯(19.30g)的二氯甲烷(200mL)溶液,滴加完畢後反應液緩慢升至30℃,繼續反應2h;反應結束後,真空旋蒸掉二氯甲烷,用去離子水洗滌,乾燥得到基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯,產率為91.2%。
3)改性雙馬來醯亞胺樹脂的製備
在20℃下,將25g(69.75 mmol) N, N』-4,4』-二苯甲烷雙馬來醯亞胺、25g(69.75 mmol) N, N』-4,4』-二苯醚雙馬來醯亞胺和35.5g(76.73 mmol)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯混合,在溫度為130℃的恆溫條件下預聚反應30min;將所得到的預聚體倒入預熱好的模具中,在145℃的真空烘箱中抽氣30min, 再以150℃/2h+180℃/2h+200℃/2h和220℃/8h工藝分別進行固化和後處理,自然冷卻後脫模,即得固化的改性雙馬來醯亞胺樹脂。
實施例12
1)2,5呋喃二甲醯氯的製備
將31.20g 2,5呋喃二甲酸、59.48g二氯亞碸和N,N-二甲基甲醯胺(DMF,催化劑,0.05mL)混合,在溫度為80℃的條件下攪拌反應5h,自然冷卻至室溫,真空旋蒸掉二氯亞碸,乾燥後得到2,5呋喃二甲醯氯,產率為99.6%。。
2)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的製備
將32.84g丁香酚和27.33g三乙胺作為鹼溶解在300mL二氯甲烷中攪拌,在-2.5±1℃下滴入2,5呋喃二甲醯氯(19.30g)的二氯甲烷(300mL)溶液,滴加完畢後反應液緩慢升至30oC,繼續反應3h;反應結束後,真空旋蒸掉二氯甲烷,用去離子水洗滌,乾燥得到基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯,產率為91.8%。
3)改性雙馬來醯亞胺樹脂的製備
在25℃下,將25g(69.75 mmol) N, N』-4,4』-二苯甲烷雙馬來醯亞胺、25g(69.75 mmol) N, N』-4,4』-二苯醚雙馬來醯亞胺和44.0g(97.76 mmol)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯混合,在溫度為140℃的恆溫條件下預聚反應30min;將所得到的預聚體倒入預熱好的模具中,在145℃的真空烘箱中抽氣30min, 再以150℃/2h+180℃/2h+200℃/2h和250℃/5h工藝分別進行固化和後處理,自然冷卻後脫模,即得固化的改性雙馬來醯亞胺樹脂。
實施例13
1)2,5呋喃二甲醯氯的製備
將31.20g 2,5呋喃二甲酸、59.48g二氯亞碸和N,N-二甲基甲醯胺(DMF,催化劑,0.05mL)混合,在溫度為80℃的條件下攪拌反應5h,自然冷卻至室溫,真空旋蒸掉二氯亞碸,乾燥後得到2,5呋喃二甲醯氯,產率為99.6%。。
2)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的製備
將34.48g丁香酚和30.36g三乙胺作為鹼溶解在500mL二氯甲烷中攪拌,在-1±1℃下滴入2,5呋喃二甲醯氯(19.30g)的二氯甲烷(500mL)溶液,滴加完畢後反應液緩慢升至30℃,繼續反應4h;反應結束後,真空旋蒸掉二氯甲烷,用去離子水洗滌,乾燥得到基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯,產率為90.6%。
3)改性雙馬來醯亞胺樹脂的製備
在30℃下,將25g(69.75 mmol) N, N』-4,4』-二苯甲烷雙馬來醯亞胺、25g(69.75 mmol)N, N』-4,4』-二苯醚雙馬來醯亞胺和54.0g(120.0 mmol)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯混合,在溫度為145℃的恆溫條件下預聚反應30min;將所得到的預聚體倒入預熱好的模具中,在145℃的真空烘箱中抽氣30min, 再以150℃/2h+180℃/2h+200℃/2h+220℃/2h和240℃/4h工藝分別進行固化和後處理,自然冷卻後脫模,即得固化的改性雙馬來醯亞胺樹脂。
實施例14
1)2,5呋喃二甲醯氯的製備
將31.20g 2,5呋喃二甲酸、59.48g二氯亞碸和N,N-二甲基甲醯胺(DMF,催化劑,0.05mL)混合,在溫度為80℃的條件下攪拌反應5h,自然冷卻至室溫,真空旋蒸掉二氯亞碸,乾燥後得到2,5呋喃二甲醯氯,產率為99.6%。
2)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的製備
將31.20g丁香酚和31.03g N-乙基二異丙胺為鹼溶解在200mL二氯甲烷中攪拌,在-4±1℃下滴入2,5呋喃二甲醯氯(19.30g)的二氯甲烷(200mL)溶液,滴加完畢後反應液緩慢升至30℃,繼續反應2h;反應結束後,真空旋蒸掉二氯甲烷,用去離子水洗滌,乾燥得到基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯,產率為91.0%。
3)改性雙馬來醯亞胺樹脂的製備
在20℃下,將25g(69.75 mmol) N, N』-4,4』-二苯甲烷雙馬來醯亞胺、25g(69.75 mmol)N, N』-4,4』-二苯醚雙馬來醯亞胺和62.5g(139.5 mmol)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯混合,在溫度為140℃的恆溫條件下預聚反應30min;將所得到的預聚體倒入預熱好的模具中,在145℃的真空烘箱中抽氣30min, 再以150℃/2h+180℃/2h+200℃/2h和220℃/8h工藝分別進行固化和後處理,自然冷卻後脫模,即得固化的改性雙馬來醯亞胺樹脂。
實施例15
1)2,5呋喃二甲醯氯的製備
將31.20g 2,5呋喃二甲酸、59.48g二氯亞碸和N,N-二甲基甲醯胺(DMF,催化劑,0.05mL)混合,在溫度為80℃的條件下攪拌反應5h,自然冷卻至室溫,真空旋蒸掉二氯亞碸,乾燥後得到2,5呋喃二甲醯氯,產率為99.6%。。
2)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的製備
將34.48g丁香酚和23.73g吡啶作為鹼溶解在500mL二氯甲烷中攪拌,在-1±1℃下滴入2,5呋喃二甲醯氯(19.30g)的二氯甲烷(500mL)溶液,滴加完畢後反應液緩慢升至30℃,繼續反應4h;反應結束後,真空旋蒸掉二氯甲烷,用去離子水洗滌,乾燥得到基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯,產率為90.8%。
3)改性雙馬來醯亞胺樹脂的製備
在30℃下,將25g(69.75 mmol) N, N』-4,4』-二苯甲烷雙馬來醯亞胺、25g(69.75 mmol) N, N』-4,4』-二苯醚雙馬來醯亞胺和75.0g(167.4 mmol)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯混合,在溫度為145℃的恆溫條件下預聚反應30min;將所得到的預聚體倒入預熱好的模具中,在145℃的真空烘箱中抽氣30min, 再以150℃/2h+180℃/2h+200℃/2h和250℃/5h工藝分別進行固化和後處理,自然冷卻後脫模,即得固化的改性雙馬來醯亞胺樹脂。
實施例16
1)2,5呋喃二甲醯氯的製備
將31.20g 2,5呋喃二甲酸、35.69g二氯亞碸和N,N-二甲基甲醯胺(DMF,催化劑,0.05mL)混合,在溫度為80℃的條件下攪拌反應2h,自然冷卻至室溫,真空旋蒸掉二氯亞碸,乾燥後,得到2,5呋喃二甲醯氯,產率為99.5%。。
2)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯的製備
將31.20g丁香酚和8.10g三乙胺、10.34g N-乙基二異丙胺和7.91g吡啶作為鹼溶解在200mL二氯甲烷中攪拌,在-5~0oC下滴入2,5呋喃二甲醯氯(19.30g)的二氯甲烷(200mL)溶液,滴加完畢後反應液緩慢升至30℃,繼續反應2h;反應結束後,真空旋蒸掉二氯甲烷,用去離子水洗滌,乾燥得到基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯,產率為91.5%。
3)改性雙馬來醯亞胺樹脂的製備
在20℃下,將50.0g(139.5 mmol)N, N』-4,4』-二苯甲烷雙馬來醯亞胺和35.5g(76.73 mmol)基於全生物質的二(4-烯丙基-2-甲氧基苯基)呋喃-2,5-二羧酸酯混合,在溫度為145℃的恆溫條件下預聚反應30min;將所得到的預聚體倒入預熱好的模具中,在145℃的真空烘箱中抽氣30min, 再以150℃/2h+180℃/2h+200℃/2h和250℃/5h工藝分別進行固化和後處理,自然冷卻後脫模,即得固化的改性雙馬來醯亞胺樹脂。