組合物、化合物及其製備方法與流程
2023-06-03 08:18:46 2
本發明涉及液晶化合物領域,尤其涉及一種化合物及其製備方法,和包括該化合物的組合物。
背景技術:
近年,液晶顯示領域的市場已非常巨大,技術亦日趨成熟,但同時對顯示質量的要求也日趨提高,尤其要求實現快速響應和降低驅動電壓以降低功耗。液晶材料為液晶顯示器重要的光電子材料之一,對改善液晶顯示器性能起著重要作用,因此液晶材料得到了巨大的發展並出現了大量液晶化合物,從聯苯腈、酯類、含氧雜環類、嘧啶環類液晶化合物發展到環己基苯類、苯乙炔類、乙基橋鍵類、端烯基液晶和各種含氟芳環類液晶化合物等,不斷滿足TN、STN和TFT-LCD等顯示性能要求。
用於顯示的液晶材料需具有適當溫度範圍、較寬液晶態溫度、較高穩定性、適合粘度以及對電場有較快響應速度,另外還需具有較寬的向列相、較低的雙折射率、非常高的電阻率、良好的抗紫外線性能、高電荷保持率以及低蒸汽壓等性能。目前,沒有任何一種液晶單體無需與其它化合物組合即能應用在液晶顯示器中;若兩種或兩種以上液晶單體混合,則可持續不斷地改變液晶的各類性質。
現有液晶材料的化合物單體一般存在具有高旋轉粘度γ1和/或較低的各向異性的缺陷,因此將其作為液晶組合物中的組分會升高組合物粘度和/或降低組合物響應速度,從而影響液晶材料的顯示質量。另外現有液晶化合物合成方法也存在著工藝繁雜、不易工業化生產、收率低以及生產成本高等問題。
技術實現要素:
本發明提供一種化合物以及包括該化合物的組合物用以解決現有技術中存在液晶化合物和組合物具有高旋轉粘度γ1和/或較低的各向異性的缺陷問題;提供一種化合物的製備方法用以解決現有技術中存在的合成工藝繁雜、不適合工業化生產、收率低的問題。
根據本發明的一方面,提供了一種化合物,該化合物如式I所示:
其中,
L1、L2和L3各自獨立地選自:單鍵、-C≡C-、-CF2O-、-OCF2-、-OCH2-、-CH2O-、-CF2CH2-、-CH2CF2-、-CH2CH2-和-(CH2)4-;
R1和R2各自獨立地選自:氫原子、具有1-15個碳原子的烷基和具有1-15個碳原子的烷氧基;
A1、A2、A3和A4各自獨立選自下述基團中的一種:
a、b和c各自獨立地選自:0、1、2和3,且a+c+e≤5。
可選地,根據本發明的化合物,所述烷基和/或所述烷氧基中的一個和/或 多個-CH2-各自獨立地被-CH=CH-、-C≡C-、-COO-、-OOC-、環丁烷或-O-替代。
可選地,根據本發明的化合物,所述烷基或所述烷氧基中的一個或多個氫各自獨立地被Cl、F、CN、OCN、OCF3、CF3、CHF2、OCHF2、SCN、NCS或SF5取代。
可選地,根據本發明的化合物,該化合物如式Ⅱ所示:
其中,A1選自下述基團中的一種:
可選地,根據本發明的化合物,該化合物如式Ⅲ所示:
其中,a為0或1;
A1選自下述基團中的一種:
A2選自下述基團中的一種:
可選地,根據本發明的化合物,該化合物如式Ⅳ所示:
可選地,根據本發明的化合物,其中,
R1選自下述基團中的一種:氫、碳原子數為1-10的直鏈烷基或碳原子數為1-10的直鏈烷氧基;
R2選自下述基團中的一種:氫、Cl、F、CN、OCF3、CF3,、SCN、CHF2、OCHF2、碳原子數為1-10的直鏈烷基或碳原子數為1-10的直鏈烷氧基;
-(F)表示苯環上有氟原子取代基或為氫。
根據本發明的另一方面,提供了一種化合物的製備方法,該方法包括:
上二氟溴甲基步驟:化合物A與二氟二溴甲烷反應,轉化為化合物B;
形成-CF2O-步驟:所述化合物B與化合物C反應,轉化為式Ⅱ所示化合物;
其中,所述化合物A、B和C如下所示:
可選地,根據本發明的製備方法,所述上二氟溴甲基步驟是在鎂存在下發生的。
可選地,根據本發明的製備方法,所述化合物A與所述鎂的當量比為1:1.1~1.3。
可選地,根據本發明的製備方法,所述形成-CF2O-步驟為化合物B和化合物C在鹼性條件下加熱至65~70℃進行反應。
根據本發明的另一方面,提供了一種化合物的製備方法,該方法包括:
上二氟溴甲基步驟:化合物D與二氟二溴甲烷反應,轉化為化合物E;
形成-CF2O-步驟:所述化合物E與化合物C反應,轉化為式Ⅳ所示化合物;
其中,所述化合物D、E和C如下所示:
可選地,根據本發明的製備方法,所述上二氟溴甲基步驟為化合物D與二氟二溴甲烷在丁基鋰存在下在-55~-70℃發生反應。
可選地,根據本發明的製備方法,所述形成-CF2O-步驟為化合物E和化合物C在鹼性環境65~70℃條件下進行反應。
根據本發明的另一方面,提供了一種組合物,該組合物包括以重量百分比計的1~90%如權利要求1-7任一所述化合物。
可選地,根據本發明的組合物,該組合物包括1~5種根據本發明的化合物單體。
本發明有益效果如下:
根據本發明的化合物具有低旋轉粘度γ1和高各向異性的特性。
根據本發明的組合物,由於包括根據本發明的化合物,同樣具有低旋轉粘度γ1和高各向異性,使組合物具有較合適的粘度和快速的響應速度,從而使液晶材料獲得良好的顯示性能,將液晶材料用於液晶顯示裝置實現快速響應和降低驅動電壓以降低功耗。
根據本發明的化合物製備方法,合成路線簡單、反應條件溫和、可控性強、後處理步驟簡單,收率高且適合工業化生產。
附圖說明
圖1為實施例3製備得到的化合物的1HNMR譜圖;
圖2為實施例6製備得到的化合物的1HNMR譜圖;
圖3為實施例9製備得到的化合物的1HNMR譜圖。
具體實施方式
具體的實施方式僅為對本發明的說明,而不構成對本發明內容的限制,下面將結合附圖和具體的實施方式對本發明進行進一步說明和描述。
根據本發明的化合物,該化合物如式I所示:
其中,
L1、L2和L3各自獨立地選自:單鍵、-C≡C-、-CF2O-、-OCF2-、-OCH2-、-CH2O-、-CF2CH2-、-CH2CF2-、-CH2CH2-和-(CH2)4-;
R1和R2各自獨立地選自:氫原子、具有1-15個碳原子的烷基和具有1-15個碳原子的烷氧基;
A1、A2、A3和A4各自獨立選自下述基團中的一種:
a、b和c各自獨立地選自:0、1、2和3,且a+c+e≤5。
根據本發明的化合物,含有四氟環己二烯結構,此結構具有不對稱的 碳原子,能誘導產生大的自發極化值,提高了該化合物的介電各向異性△ε,其作為組合物的一種組分時使組合物具有快響應速度,易於產生良好的相序。該化合物的結構穩定,具有寬液晶態溫度範圍,具有較好的低溫互溶性,其作為組合物應用於光學器件時,使光學器件實現較低的閾值電壓。另外該化合物具有低旋轉粘度γ1可以改善液晶組合物材料和顯示器性能,對實現顯示器的快速響應具有重要意義。含有此類化合物的液晶組合物可應用於製備驅動電壓低,溫度範圍寬、響應速度快的液晶顯示裝置。其中a+c+e≤5,是為了限制基團的個數,基團個數太多,分子量增大,使其不易混合,不易和其他單體形成組合物,不利於應用。L1、L2和L3選自:單鍵、-C≡C-、-CF2O-、-OCF2-、-OCH2-、-CH2O-、-CF2CH2-、-CH2CF2-、-CH2CH2-和-(CH2)4-,進一步地優選-CF2O-和-OCF2-,含有該橋鍵會使化合物的向列相溫度範圍很大程度的擴大,同時旋轉粘度γ1有所降低,另外由於所述基團的偶極矩的貢獻,端基氟原子的偶極矩在一定程度上提高,從而使液晶分子的介電各向異性有所增加。
根據本發明化合物的一種表現形式,烷基和/或烷氧基中的一個和/或多個-CH2-各自獨立地被-CH=CH-、-C≡C-、-COO-、-OOC-、環丁烷或-O-替代。
根據本發明化合物的一種表現形式,烷基或烷氧基中的一個或多個氫各自獨立地被Cl、F、CN、OCN、OCF3、CF3、CHF2、OCHF2、SCN、NCS或SF5取代。
根據本發明化合物的一種表現形式,該化合物優選:
其中,A1選自下述基團中的一種:
根據本發明化合物的一種表現形式,該化合物進一步優選下列化合物:
根據本發明化合物的一種表現形式,該化合物優選:
其中,a為0或1;
A1選自下述基團中的一種:
A2選自下述基團中的一種:
進一步地優選:
根據本發明化合物的一種表現形式,該化合物優選:
進一步地優選:
根據本發明化合物的一種表現形式,其中,R1選自下述基團中的一種:氫、碳原子數為1-10的直鏈烷基或碳原子數為1-10的直鏈烷氧基;R2選自下述基團中的一種:氫、Cl、F、CN、OCF3、CF3,、SCN、CHF2、OCHF2、碳原子數為1-10的直鏈烷基或碳原子數為1-10的直鏈烷氧基;-(F)表示苯環上有氟原子取代基或為氫。
根據本發明的化合物具有低旋轉粘度γ1和高各向異性。
根據本發明化合物的製備方法,該方法包括:上二氟溴甲基步驟:化合物A與二氟二溴甲烷反應,轉化為化合物B;形成-CF2O-步驟:所述化合物B與化合物C反應,轉化為式Ⅱ所示的化合物;其中,所述化合物A、B和C如下所示:
其中,在上二氟溴甲基步驟優選在鎂存在下發生的,化合物A與鎂的當量比優選1:1.1~1.3,進一步地優選1:1.2;形成-CF2O-步驟優選化合物B和化合物C在鹼性條件下加熱至65~70進行反應。
根據本發明化合物的製備方法,上二氟溴甲基的具體操作如下:將1eq化合物A和1.1~1.3eq的鎂和適量的碘加入溶劑中,滴加二氟二溴甲烷,在-40℃~-70℃反應。其中溶劑優選四氫呋喃(THF)。其中化合物A和鎂的加料順序可以根據不同化合物的製備方法不同。如其中的一種加料順序為:可以先將鎂加入四氫呋喃的溶劑中,加入適量的碘,加入部分化合物A,待反應引發後,緩慢加入剩餘的化合物A,攪拌一段時間,優選30~90min,後再滴加二氟二溴甲 烷;另一種加料順序為:化合物A和鎂以及適量的碘同時加入THF中,加熱引發回流30~90min後,降溫至-40℃~-70℃滴加二氟二溴甲烷。該步驟的後處理具體操作為:加入水或鹽酸水溶液淬滅、萃取,水洗、乾燥濃縮得粗品。將粗品用無水乙醇重結晶,得到化合物B。收率能達到70%以上,氣相色譜檢測其純度能達99.6%以上。
根據本發明化合物的製備方法,形成-CF2O-步驟的具體操作如下:將1eq化合物B、1.1~1.3eq的化合物C及1.8~2.2eq的鹼加入溶劑中,所述鹼優選無水碳酸鉀,所述溶劑優選DMSO,攪拌加熱至65~70℃反應,所述化合物C優選1.2eq,所述鹼優選2eq。該步驟的後處理一般為降至室溫後過濾,濾除固體,在濾液中加水,對濾液進行萃取,優選用二氯甲烷進行萃取,將有機相水洗、乾燥,濃縮,過柱,重結晶得到目標化合物。
根據本發明的一種化合物的製備方法,該方法包括:
上二氟溴甲基步驟:化合物D與二氟二溴甲烷反應,轉化為化合物E;
形成-CF2O-步驟:所述化合物E與化合物C反應,轉化為式Ⅳ所示化合物;
其中,所述化合物D、E和C如下所示:
其中,上二氟溴甲基步驟為化合物D與二氟二溴甲烷在丁基鋰存在在-55~-70℃發生反應;形成-CF2O-步驟為化合物E和化合物C在鹼性環境65~70℃條件下進行反應。
根據本發明化合物的製備方法,其中上二氟溴甲基步驟具體操作為:將1eq的化合物D溶解在溶劑中,溶劑優選四氫呋喃(THF),通氮氣保護,降溫至-55~-70℃,在此溫度滴加丁基鋰,滴畢攪拌50~70min,滴加二氟二溴甲烷,滴加完畢保溫反應。該步驟的後處理為:反應完畢升至室溫,加水或鹽酸水溶液淬滅,萃取,水洗、乾燥濃縮得化合物E的粗品。
根據本發明化合物的製備方法,形成-CF2O-步驟的具體操作如下:將1eq化合物E、1.1~1.3eq的化合物C及1.8~2.2eq的鹼加入溶劑中,所述鹼優選無水碳酸鉀,所述溶劑優選DMSO,攪拌加熱至65~70℃反應,所述化合物C優選1.2eq,所述鹼優選2eq。該步驟的後處理一般為降至室溫後過濾,濾除固體,在濾液中加水,對濾液進行萃取,優選用二氯甲烷進行萃取,將有機相水洗、乾燥,濃縮,過柱,重結晶得到目標化合物。
根據本發明的化合物製備方法,合成路線簡單、反應條件溫和、可控性強、後處理步驟簡單,收率高且適合工業化生產。
根據本發明的組合物,該組合物包括以重量百分比計的1~90%根據本發明的化合物,進一步地優選5~60%,更進一步地優選10-40%。該組合物優選包括1~5種根據本發明化合物的單體。
根據本發明的組合物,由於包括根據本發明的化合物,同樣具有低旋轉粘度γ1和高介電各向異性△ε,使組合物具有較合適的粘度和快速的響應速度,從而使液晶材料獲得良好的顯示性能,將液晶材料用於液晶顯示裝置實現快速響應和降低驅動電壓以降低功耗。
綜上所述,根據本發明的化合物及其製備方法可選因素較多,根據權利要求可以組合出不同的實施例。實施例僅用於對本發明進行說明,並不對本發明構成限制。下面將結合附圖對本發明進行進一步描述。
由於根據本發明化合物中的取代基的多樣性,權利要求的組合結果也眾多,因此選用具體化合物的合成方法作為實施例對本發明的製備方法進行說明。
根據本發明製備式Ⅱ所示化合物的方法,選用式Ⅱ代表的化合物中的一種具體化合物進行描述,該具體化合物如式Ⅱa-1所示:
製備式Ⅱa-1所示具體化合物中上二氟溴甲基步驟的化學反應方程式如下所示:
形成-CF2O-步驟的化學反應方程式如下所示:
實施例1-3為製備如式Ⅱa-1所示化合物的具體方法。
實施例1
根據本發明化合物的製備方法,首先進入上二氟溴甲基步驟:將1.1eq鎂加入四氫呋喃的溶劑中,加入適量的碘(如1粒),加入0.1eq化合物A-1,待反應引發後,緩慢加入0.9eq化合物A-1,攪拌30min後,在-65℃滴加1.4eq二氟二溴甲烷後進行反應,反應完成後進行後處理,升至室溫後加入水或鹽酸水溶液淬滅、萃取,水洗、乾燥濃縮得化合物B-1,收率達70%以上;然後進入形成-CF2O-步驟:將1eq化合物B-1、1.1eq的化合物C-1及1.8eq的無水碳酸鉀加入DMSO中,攪拌加熱至65℃反應,反應完成後降至室溫過濾,濾除固體,在濾液中加水,用二氯甲烷進行萃取,將有機相水洗、乾燥,濃縮,過柱,重結晶得到目標化合物Ⅱa-1。
實施例2
根據本發明化合物的製備方法,首先進入上二氟溴甲基步驟:將1.3eq鎂加入四氫呋喃的溶劑中,加入適量的碘(如1粒),加入0.3eq化合物A-1,待反應引發後,緩慢加入0.7eq的化合物A-1,攪拌90min,在-70℃滴加1.6eq二氟二溴甲烷,反應完畢後,加水或鹽酸水溶液淬滅、萃取,水洗、乾燥濃縮得化 合物B-1,收率能達到70%以上;然後進入形成-CF2O-步驟:將1eq化合物B-1、1.3eq的化合物C-1及2.2eq的無水碳酸鉀加入DMSO中,攪拌加熱至70℃反應,反應完成後降至室溫過濾,濾除固體,在濾液中加水,用二氯甲烷對濾液進行萃取,將有機相水洗、乾燥,濃縮,過柱,重結晶得到目標化合物Ⅱa-1。
實施例3
根據本發明化合物的製備方法,首先進入上二氟溴甲基步驟:將1.2eq鎂加入四氫呋喃的溶劑中,加入適量的碘(如1粒),加入0.2eq化合物A-1,待反應引發後,緩慢加入0.8eq的化合物A-1,攪拌60min,後再滴加1.5eq二氟二溴甲烷,反應完成後加入水或鹽酸水溶液淬滅、萃取,水洗、乾燥濃縮得化合物B-1,收率能達到70%以上;然後進入形成-CF2O-步驟:將1eq化合物B-1、1.2eq的化合物C及2.0eq的無水碳酸鉀加入DMSO中,攪拌加熱至68℃反應,反應完成後降至室溫後過濾,濾除固體,在濾液中加水,對濾液進行萃取,優選用二氯甲烷進行萃取,將有機相水洗、乾燥,濃縮,過柱,重結晶得到目標化合物Ⅱa-1。
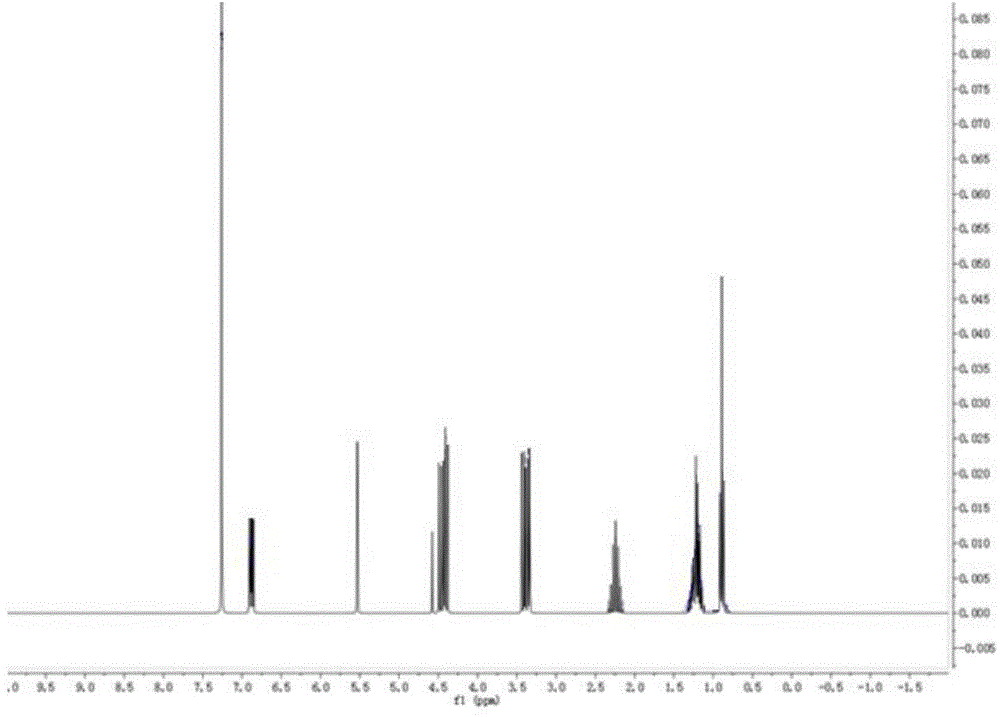
對實施例3製備得到的化合物進行了核磁表徵,譜圖如圖1所示,具體譜圖解析為:1HNMR(500MHz,CDCl3)δ6.86(ddt,J=12.8,10.1,1.6Hz,2H),4.31(t,J=41.8Hz,1H),2.85–2.61(m,1H),1.97–1.59(m,4H),1.41–0.76(m,3H);由此可見製備得到的化合物為化合物Ⅱa-1。
經測試,根據實施例3製備得到的化合物Ⅱa-1的MP為45℃,△ε為22.3,△n為0.129。
根據本發明製備式Ⅱ所示化合物的方法,選用式Ⅱ代表的化合物中的另一種具體化合物進行描述,該具體化合物如式Ⅱc-1所示:
製備式Ⅱc-1所示具體化合物中上二氟溴甲基步驟的化學反應方程式如下 所示:
形成-CF2O-步驟的化學反應方程式如下所示:
另外原料化合物A-2的製備的化學反應方程式如下所示:
首先合成原料A-2的操作為:將1eq的化合物H和1eq的2-丙基-1,3-丙二醇加入乾燥的甲苯溶劑中,加入適量的對甲苯磺酸,加熱回流4小時,分出生成的水,冷卻至室溫,水洗甲苯溶液,然後將甲苯溶液濃縮得化合物A-2,收率為95%,氣相色譜的純度98%。
現將製備得到的原料A-2用於實施例4-6中,用於合成式Ⅱc-1所示的化合物。
實施例4
根據本發明化合物的製備方法,首先進入上二氟溴甲基步驟:將1eq化合物A-2和1.1eq的鎂和適量的碘加入四氫呋喃中,緩慢加熱引發後,回流50min,降溫至-40℃,滴加1.1eq的二氟二溴甲烷,在此溫度下反應,反應完成後加水淬滅、萃取,水洗、乾燥濃縮得粗品。將粗品用無水乙醇重結晶,得到化合物B-2,收率為75%,氣相色譜檢測其純度能達99.6%以上;然後進入形成-CF2O-步驟:將1eq化合物B-2、1.1的化合物C-1及1.8eq的無水碳酸鉀加入DMSO中,攪拌加熱至65℃反應,反應完成後降至室溫後過濾,濾除固體,在濾液中加水,對濾液用二氯甲烷進行萃取,將有機相水洗、乾燥,濃縮,過柱,重結晶得到目標化合物Ⅱc-1。
實施例5
根據本發明化合物的製備方法,首先進入上二氟溴甲基步驟:將1eq化合物A-2和1.3eq的鎂和適量的碘加入四氫呋喃中,緩慢加熱引發後,回流70min,降溫至-50℃,滴加1.1eq的二氟二溴甲烷,在此溫度下反應,反應完成後加水淬滅、萃取,水洗、乾燥濃縮得粗品。將粗品用無水乙醇重結晶,得到化合物B-2,收率為78%,氣相色譜檢測其純度能達99.8%以上;然後進入形成-CF2O-步驟:將1eq化合物B-2、1.3的化合物C-1及2.2eq的無水碳酸鉀加入DMSO中,攪拌加熱至70℃反應,反應完成後降至室溫後過濾,濾除固體,在濾液中加水,對濾液用二氯甲烷進行萃取,將有機相水洗、乾燥,濃縮,過柱,重結晶得到目標化合物Ⅱc-1。
實施例6
根據本發明化合物的製備方法,首先進入上二氟溴甲基步驟:將1eq化合物A-2和1.2eq的鎂和適量的碘加入四氫呋喃中,緩慢加熱引發後,回流60min,降溫至-45℃,滴加1.2eq的二氟二溴甲烷,在此溫度下反應,反應完成後加水淬滅、萃取,水洗、乾燥濃縮得粗品。將粗品用無水乙醇重結晶,得到化合物B-2,收率為80%,氣相色譜檢測其純度能達99.7%以上;然後進入形成-CF2O-步驟:將1eq化合物B-2、1.1的化合物C-1及2.0eq的無水碳酸鉀加入DMSO中,攪拌加熱至68℃反應,反應完成後降至室溫後過濾,濾除固體,在濾液中加水,對濾液用二氯甲烷進行萃取,將有機相水洗、乾燥,濃縮,過柱,重結晶得到目標化合物Ⅱc-1。
對實施例6製備得到的化合物進行了核磁表徵,譜圖如圖2所示,具體譜圖解析為:1H NMR(500MHz,CDCl3)δ6.87(ddt,J=10.1,2.7,1.6Hz,2H),5.34(d,J=3.3Hz,1H),4.62–4.09(m,3H),3.56–3.19(m,3H),2.22–1.85(m,1H),1.55–1.06(m,4H),1.03–0.72(m,3H);由此可見製備得到的化合物為化合物Ⅱc-1。
根據實施例6製備得到的化合物Ⅱc-1的MP為60℃,△ε:19.8,△n:0.198。
根據本發明的化合物式Ⅳ的製備方法,選用式Ⅳ代表的化合物中的一種具體化合物進行描述,該具體化合物如式Ⅲb-1所示:
上二氟溴甲基步驟的化學反應方程式如下所示:
形成-CF2O-步驟的化學反應方程式如下所示:
其中原料化合物D-1合成的化學方程式如下所示:
其中原料化合物D-1的合成操作為,將1eq的化合物G與1.2eq的3,5-二氟溴苯,3eq的氟化銫加入甲苯、乙醇和水的體積比為4:3:3的混合溶劑中,氮氣保護,加入催化劑量的四(三苯基膦)鈀,攪拌加熱至回流反應,反應完畢後降溫,分液,用甲苯萃取,將有機相合併水洗至中性,蒸乾溶劑,將所得物溶於甲苯過矽膠柱脫色,用甲苯洗脫,得到白色固體D-1。收率大於91%,氣相色譜純度為99.2%。
將上述製備得到的化合物D-1用於實施例7-9中,用於製備化合物式式Ⅲb-1。
實施例7
根據本發明化合物的製備方法,首先進入上二氟溴甲基步驟具體操作為:將1eq的化合物D-1溶解在四氫呋喃中,通氮氣保護,降溫至-55℃,在此溫度滴加丁基鋰,滴畢攪拌50min,滴加二氟二溴甲烷,滴加完畢保溫反應。該步驟的後處理為:反應完畢升至室溫,加水或鹽酸水溶液淬滅,萃取,水洗、乾燥濃縮得化合物E-1的粗品。
然後進入形成-CF2O-步驟:將1eq化合物E-1、1.1eq的化合物C-1及1.8eq的無水碳酸鉀加入DMSO中,攪拌加熱至65℃反應。該步驟的後處理一般為降至室溫後過濾,濾除固體,在濾液中加水,對濾液進行萃取,優選用二氯甲烷進行萃取,將有機相水洗、乾燥,濃縮,過柱,重結晶得到目標化合物式Ⅲb-1。
實施例8
根據本發明化合物的製備方法,首先進入上二氟溴甲基步驟具:將1eq的化合物D-1溶解在四氫呋喃中,通氮氣保護,降溫至-70℃,在此溫度滴加丁基鋰,滴畢攪拌70min,滴加二氟二溴甲烷,滴加完畢保溫反應。該步驟的後處理為:反應完畢升至室溫,加水或鹽酸水溶液淬滅,萃取,水洗、乾燥濃縮得化合物E-1的粗品。
然後進入形成-CF2O-步驟:將1eq化合物E、1.3eq的化合物C及2.2eq的無水碳酸鉀加入DMSO中,攪拌加熱至70℃反應。該步驟的後處理為降至室溫後過濾,濾除固體,在濾液中加水,對濾液進行萃取,優選用二氯甲烷進行萃取,將有機相水洗、乾燥,濃縮,過柱,重結晶得到目標化合物式式Ⅲb-1。
實施例9
根據本發明化合物的製備方法,首先進入上二氟溴甲基步驟:將1eq的化合物D-1溶解在四氫呋喃中,通氮氣保護,降溫至-60℃,在此溫度滴加丁基鋰,滴畢攪拌60min,滴加二氟二溴甲烷,滴加完畢保溫反應。該步驟的後處理為:反應完畢升至室溫,加水或鹽酸水溶液淬滅,萃取,水洗、乾燥濃縮得 化合物E-1的粗品。
然後進入形成-CF2O-步驟:將1eq化合物E-1、1.2eq的化合物C-1及2.0eq的無水碳酸鉀加入DMSO中,攪拌加熱至68℃反應。該步驟的後處理一般為降至室溫後過濾,濾除固體,在濾液中加水,對濾液進行萃取,優選用二氯甲烷進行萃取,將有機相水洗、乾燥,濃縮,過柱,重結晶得到目標化合物式Ⅲb-1。
對實施例9製備得到的化合物進行了核磁表徵,譜圖如圖3所示,具體譜圖解析為:H NMR(500MHz,CDCl3)δ7.20–6.96(m,6H),6.88(ddt,J=12.8,10.1,1.6Hz,2H),4.94(d,J=13.4Hz,2H),2.61(t,J=12.3Hz,2H),1.73–1.49(m,2H),0.94(t,J=13.2Hz,3H);由此可見製備得到的化合物為化合物式Ⅲb-1。
根據實施例9製備得到的化合物式Ⅲb-1的MP為46.5℃,△ε:17.0,△n:0.188。
根據實施例1-9的的製備方法均製備出了根據本發明的化合物,合成路線簡單、反應條件溫和、可控性強、後處理步驟簡單,收率高且適合工業化生產。
另外,根據本發明的化合物可以與其他化合物組成組合物,該組合物包括以重量百分比計的1~90%根據本發明的化合物,進一步地優選5~60%,更進一步地優選10-40%。該組合物優選包括1~5種根據本發明化合物的單體,更進一步地優選包括2-4種。實施例10-12進一步地對本發明的組合物進行描述。
實施例10
根據本發明的組合物包括以重量百分比計的1%的式Ⅰ所示化合物,該組合物包括1種式Ⅰ所示化合物的單體。
實施例11
根據本發明的組合物包括以重量百分比計的90%的式Ⅰ所示化合物,該組合物包括5種式Ⅰ所示化合物的單體。
實施例12
根據本發明的組合物包括以重量百分比計的5%的式Ⅰ所示化合物,該組 合物包括2種式Ⅰ所示化合物的單體。
實施例13
根據本發明的組合物包括以重量百分比計的60%的式Ⅰ所示化合物,該組合物包括4種式Ⅰ所示化合物的單體。
在實施例10-13的組合物中包括本發明中的化合物,剩餘部分為母體液晶。所述母體液晶優選由表1物質按下述重量百分比組成。
表1
另外,在本發明中對製備得到的化合物和組合物進行了性能測試。其中,MP表示熔點、△ε表示介電各向異性、△n表示光學各向異性。
對化合物的性能測定存在兩種情況:一是將化合物本身作為試樣,二是將化合物與母液晶混合來作為試樣。
將化合物與母液晶混合進行測定時:將15wt%的式Ⅰ所示的液晶化合物與85wt%的母液晶混合來製作試樣,用所得試樣進行測定,所得測定值按下式所示的外推法來計算外推值:外推值=[100×(試樣測定值)-(母液晶的重量百分率)×(母液晶的測定值)]/化合物的重量百分率。
液晶化合物的物理性質測定方法按照本行業的規範進行,參見《液晶器件手冊》航空工業出版社出版測定值中,當以液晶化合物本身作為試樣時,記錄所得值作為實驗值,當以液晶化合物與母液晶的混合物作為試樣時,記錄由外推法得到的值作為實驗值。其中對光學各向異性和介電常數各向異性的測定分別按下述條件進行:
1.光學各向異性(△n):在25℃,利用阿貝折射計選用589nm波長光進行測試。
沿同一方向對主稜鏡(Pri3m)表面進行摩擦後,將試樣滴加到主稜鏡上。折射率(n11)是在偏光方向與摩擦方向平行時測定,折射率(n⊥)是在偏光方向與摩擦方向垂直時測定。光學各向異性(△n)的值由△n=n11-n⊥來計算。
2.介電常數各向異性(△ε)的測定:
在25℃,用惠普公司的HP4284a測定儀進行測定,測定液晶分子在軸方向的介電常數(ε11),測定液晶分子短軸方向的介電常數(ε⊥),介電常數各向異性通過公式△ε=ε11-ε⊥來計算。
通過上述性能的測定結果,可以看出根據本發明的化合物具有低旋轉粘度 γ1和高各向異性。
顯然,本領域的技術人員可以對本發明進行各種改動和變型而不脫離本發明的精神和範圍。這樣,倘若本發明的這些修改和變型屬於本發明權利要求及其等同技術的範圍之內,則本發明也意圖包含這些改動和變型在內。