一步法原位合成Cu‑SAPO‑18分子篩催化劑的製備方法及其用途與流程
2023-05-28 15:56:22 3
本發明屬於化工及環保領域,涉及cu-sapo-18分子篩催化劑的製備方法,具體涉及一步法原位製備cu-sapo-18催化劑的方法,以及製備產物cu-sapo-18分子篩催化劑用於柴油車後處理urea-scr系統催化器中氮氧化合物淨化過程。
背景技術:
目前nh3-scr催化劑的研究熱點之一是過渡金屬負載或者離子交換的微孔分子篩催化劑。根據國際純粹與應用化學聯合會(iupac)的定義,微孔分子篩分為小孔(<0.5nm)、中孔(0.5~0.7nm)、大孔(0.7~2nm)。研究表明,與中孔、大孔分子篩相比,小孔分子篩由於其特殊的孔道結構,在nh3-scr反應中具有高催化活性、高水熱穩定性、優異的抗鹼金屬中毒性和抗積碳性能。
aei型結構分子篩(ssz-39和sapo-18),具有三維八元環孔道體系,由於該結構中彼此連接雙六元環呈鏡像對稱分布,因此形成的八元環孔道結構呈直線型,其八元環孔口直徑為0.38nm×0.38nm。與cha型分子篩相比,aei型分子篩催化劑在nh3-scr反應中表現出更優異的催化性能,水熱穩定性能和抗積碳性能,作為滿足未來更高排放標準的新型小孔分子篩nh3-scr催化劑,其中以sapo-18為載體製備的cu-sapo-18催化劑受到國內外研究者的廣泛關注。
目前報導合成cu-sapo-18分子篩催化劑有兩種方法,一種方法是以sapo-18為載體,採用離子交換方法負載活性組分製備,如yangrt等利用n-n二異丙基乙胺為模板劑合成sapo-18載體,再將h-sapo-18進行與nh4no3進行離子交換製備nh4-sapo-18,然後將製備的nh4-sapo-18和一定濃度的銅鹽前驅體溶液在一定溫度下交換、過濾、洗滌、乾燥和高溫煅燒製備cu-sapo-18分子篩催化劑(qye,.etal.,appliedcatalysisa:general,2012,427–428(10):24–34)。葉青等利用n-n二異丙基乙胺為模板劑合成sapo-18載體,然後將製備的sapo-18載體和一定濃度的氯化銅和氯化鐵溶液在一定溫度下交換、過濾、洗滌、乾燥和高溫煅燒製備cu/fe-sapo-18分子篩催化劑(cn102626653b),由於sapo-18載體孔道尺寸和交換容量的限制,為了保證活性組分銅的負載量和高分散度,需要進行多次離子交換過程,該過程不僅影響分子篩載體骨架穩定性能,同時在交換過程中銅鹽前驅體溶液利用率低,洗滌過程中消耗大量純淨水,高溫煅燒過程消耗大量能源。另一種是採用一步法原位合成,corma等利用n-n二甲基-3,5-二甲基哌啶鎓和銅胺配合物合成cu-sapo-18分子篩催化劑,該路線避免了合成過程中多次煅燒、離子交換工藝,同時通過改變分子篩結構中矽原子的配位環境、調控表面酸濃度和酸強度,但是(martínez-franco,r.etal.directsynthesisdesignofcu-sapo-18,averyefficientcatalystforthescrofnoxjournalofcatalysis,2014,319:36-43)採用的n,n-二甲基-3,5-二甲基哌啶正離子作為共模板劑,價格昂貴,合成cu-sapo-18分子篩成本太高,不利於大規模的工業化生產推廣。
技術實現要素:
本發明的任務是提供一種一步法原位合成cu-sapo-18分子篩催化劑的製備方法,使其具有造價低廉、工藝簡單、綠色環保、分子篩活性組分銅的含量可控和可調控分子篩結構中矽原子的配位環境表面酸濃度和酸強度等特點,以克服現有技術的不足。
實現本發明的技術方案是:
本發明提供的一步法原位合成cu-sapo-18分子篩催化劑的製備方法,包括以下步驟:
(1)將鋁源、正磷酸、矽源、五水硫酸銅、四乙烯五胺和有機模板劑n,n-二異丙基乙胺依次加入去離子水中充分攪拌後得到初始凝膠;投料摩爾比按以下方案a、方案b、方案c或方案d進行:
方案a:投料摩爾比為:p2o5/al2o3=0.20~0.78、sio2/al2o3=0.10~0.98、硫酸銅-四乙烯五胺/al2o3=0.02~0.18、n,n-二異丙基乙胺/al2o3=0.90~1.60和h2o/al2o3=15.00~50.00;
方案b:al2o3、p2o5、sio2、硫酸銅-四乙烯五胺、n,n-二異丙基乙胺和h2o的投料摩爾比依次為1:0.2~0.78:0.1~0.98:0.02~0.18:0.90~1.60:15~50;
方案c:al2o3、p2o5、sio2、硫酸銅-四乙烯五胺、n,n-二異丙基乙胺和h2o的投料摩爾比依次為1:0.2~0.78:0.2~0.8:0.09:0.90~1.60:15~50;
方案d:al2o3、p2o5、sio2、硫酸銅-四乙烯五胺、n,n-二異丙基乙胺和h2o的投料摩爾比依次為:
1:0.2~0.78:0.1~0.98:0.02:0.90~1.60:15~50或
1:0.2~0.78:0.1~0.98:0.06:0.90~1.60:15~50或
1:0.2~0.78:0.1~0.98:0.09:0.90~1.60:15~50或
1:0.2~0.78:0.1~0.98:0.12:0.90~1.60:15~50或
1:0.2~0.78:0.1~0.98:0.15:0.90~1.60:15~50或
1:0.2~0.78:0.1~0.98:0.18:0.90~1.60:15~50或
1:0.2~0.78:0.2:0.09:0.90~1.60:15~50或
1:0.2~0.78:0.4:0.09:0.90~1.60:15~50或
1:0.2~0.78:0.6:0.09:0.90~1.60:15~50或
1:0.2~0.78:0.8:0.09:0.90~1.60:15~50;
(2)將攪拌完全的凝膠置於水熱反應釜中,在150~220℃下晶化1-7d,室溫冷卻,經離心過濾、洗滌、乾燥,在500~850℃下煅燒2~6小時,得到cu-sapo-18分子篩催化劑。
本發明上述方法中所述的鋁源為異丙醇鋁、擬水勃姆石、勃姆石的一種或幾種;所述的矽源為正矽酸四乙酯、矽溶膠和氣態sio2粉末中的一種或幾種。
本發明採用以雙模板劑一步法原位合成cu-sapo-18分子篩催化劑,通過控制硫酸銅-四乙烯五胺的投入比例可以調控銅的負載量為2.28~6.73wt.%,同時控制矽溶膠的添加量控制矽鋁比為0~0.3,得到具有高結晶度、優異的催化活性和水熱穩定性及抗鹼金屬中毒性,並且粒徑分布均勻的cu-sapo-18分子篩催化劑。
本發明提供的方法中,優選控制al2o3、p2o5、sio2、硫酸銅-四乙烯五胺、n,n-二異丙基乙胺和h2o的投料摩爾比為1:0.2~0.78:0.1~0.98:0.02~0.18:0.90~1.60:15~50,採用icp對製備的cu-sapo-18催化劑進行檢測,銅的質量分數為2.28~6.73wt.%;優選al2o3、p2o5、sio2、硫酸銅-四乙烯五胺、n,n-二異丙基乙胺和h2o的投料摩爾比為1:0.2~0.78:0.1~0.98:0.09:0.90~1.60:15~50,此時銅的質量分數為4.96±0.1wt.%。
本發明提供的方法中,優先控制al2o3、p2o5、sio2、硫酸銅-四乙烯五胺、n,n-二異丙基乙胺和h2o的投料摩爾比為1:0.2~0.78:0.1~0.98:0.09:0.90~1.60:15~50,採用xrf對製備的cu-sapo-18催化劑進行檢測,矽鋁比為0~0.3;優選al2o3、p2o5、sio2、硫酸銅-四乙烯五胺、n,n-二異丙基乙胺和h2o的投料摩爾比1:0.2~0.78:0.6:0.09:0.90~1.60:15~50,此時矽鋁比為0.04~0.08。
在本發明提供的方法中,晶化溫度為150~220℃,晶化時間為1~7d,優選晶化溫度為190℃,晶化時間為72小時。
在本發明提供的方法中,煅燒溫度為500~850℃,優選煅燒溫度為550℃;煅燒時間為2~6h,優選3小時。
本發明的又一個任務是提供一種由本發明方法製備得到的cu-sapo-18分子篩催化劑。
本發明的第三個任務是將本發明方法製備得到的cu-sapo-18分子篩催化劑用於柴油車後處理urea-scr系統催化器中氮氧化合物淨化過程。
本發明的優點在於:(1)本發明方法與現有合成cu-sapo-18的方法相比,本發明合成方法具有造價低廉、工藝簡單、綠色環保、分子篩活性組分銅的含量可控和可調控分子篩結構中矽鋁比等優點,克服了傳統一步合成法造價成本高,原料利用率低等不足;(2)採用按照不同比例混合的雙模板劑一步法原位合成cu-sapo-18,得到分子篩矽鋁比在0~0.3的範圍內可調,活性組分銅的質量分數在2.28~6.730.1wt.%範圍;(3)採用本方法製備的cu-sapo-18催化劑(銅的負載量為4.86~4.96wt.%和矽鋁比為0~0.3可調控),在高空速的條件下(30,000h-1),其新鮮活性保持90%以上的nox的轉化率的溫度區間為200~525℃,說明了該催化劑適用於柴油車後處理urea-scr系統催化器中氮氧化合物淨化過程;(4)採用本發明方法製備的cu-sapo-18催化劑在含10wt.%水蒸氣的空氣氛圍中,在800℃老化16h後,其催化劑性質穩定,仍較完整的保持aei骨架,在225~500℃保持90%以上的nox的轉化率,表現出極高的抗水熱穩定性能;(5)採用本方法製備的cu-sapo-18催化劑在不同濃度的鹼金屬中毒條件下,在225~475℃保持90%以上的nox的轉化率,表現出高的抗鹼金屬性能;
附圖說明
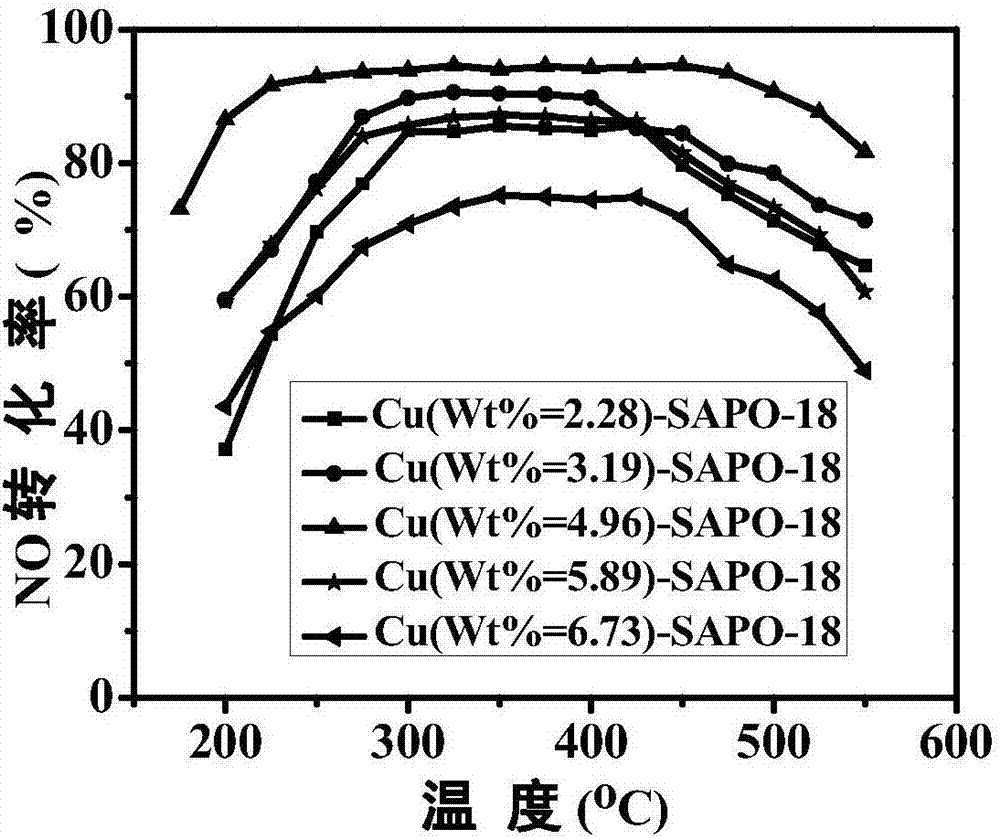
圖1是實施例1新鮮催化劑的nox轉化率評價圖;
圖2是實施例1不同條件催化劑的nox轉化率評價圖;
圖3是實施例1新鮮催化劑的xrd圖;圖1~3表明,cu-sapo-18分子篩催化劑的催化活性和骨架穩定性受不同銅負載量活性組分的影響,銅含量過高樣品在中高溫區間(450~550℃)表現出較低的nox轉化催化活性。而過低的活性組分含量的樣品在中低溫區間(200~350℃)表現出較低的nox轉化催化活性。其中,最佳銅負載量為4.96wt.%,其新鮮老化的活性均達到最佳,表明其具有優良的抗水熱性能。同時圖3中均未出現cuo的特徵峰(2θ=35.6°、39.8°),說明其合成的分子篩催化劑不存在cuo,或者cuo高度分散的;
圖4是實施例3新鮮和不同老化條件後催化劑的nox轉化率評價圖,圖4表示,cu-sapo-18分子篩催化劑在經過三種不同程度的水熱老化條件後,仍具有與新鮮催化劑相似的優異催化活性;
圖5是實施例3催化劑的抗鹼金屬性能評價圖,圖5表明,鹼土金屬中毒後的催化劑,活性仍然基本得到保持;
圖6是實施例3催化劑的sem圖,圖6表明該催化劑晶粒分布均勻;
具體實施方式
下面結合附圖並通過具體實施方式來進一步說明本發明的技術方案。
在本發明中,催化劑的評價採用如下方法:將cu-sapo-18催化劑粉末與少量水混合,製備漿液,塗覆於堇青石蜂窩陶瓷基體小樣,催化劑塗覆量約為250g·l-1,樣品在100℃乾燥2h,500℃焙燒2h,即為製備的整體式cu-sapo-18催化劑,將其放入固定床活性評價裝置中進行,模擬煙氣組成為1000ppmno,1100ppmnh3,5%o2和10%h2o,反應空速為30,000h-1。
在本發明中,催化劑的水熱老化測試採用如下方法:將製備好的整體催化劑cu-sapo-18置於管式爐中,通過程序控制器控制其達到目標溫度後,通入10%的水蒸氣,流速設定為120ml/h。達到目標時間後,關閉程序控制器,關閉水蒸氣並降至室溫即可。
實施例1
將五水硫酸銅加入去離子水在室溫下攪拌20分鐘,再向其緩慢加入等摩爾比的四乙烯五胺,攪拌3小時後,再向其加入稀釋的50%正磷酸滴加入銅胺溶液中,並持續攪拌3小時,緩慢加入勃姆石,劇烈攪拌3小時後,混合後加入矽溶膠和模板劑n,n-二異丙基乙胺和餘下的水攪拌6小時後,將攪拌完全的凝膠置於水熱反應釜中,190℃反應72小時,反應完成後冷卻至室溫,經離心過濾、去離子水洗滌、100℃乾燥12小時,於馬弗爐中550℃下煅燒4個小時,得到cu-sapo-18分子篩催化劑。在該方法中,控制各反應原料的添加量的摩爾比關係如表一所示:
表一
對五組實驗得到的cu-sapo-18分子篩催化劑,分別進行icp測試,測得活性組分銅的質量分數分別為2.28wt.%、3.19wt.%、4.96wt.%、5.89wt.%、、6.73wt.%,說明該方法能夠在較寬的範圍內,有效控制催化劑上的cu含量。
圖1~3是含有不同負載量活性組分的cu-sapo-18分子篩催化劑的水熱老化前後的催化性能和xrd圖,該圖表明,cu-sapo-18分子篩催化劑的催化活性和骨架穩定性受不同銅負載量活性組分的影響,銅含量過高樣品在中高溫區間(450~550℃)表現出較低的nox轉化催化活性。而過低的活性組分含量的樣品在中低溫區間(200~350℃)表現出較低的nox轉化催化活性。其中,最佳銅負載量為4.96wt.%,其新鮮老化的活性均達到最佳,表明其具有優良的抗水熱性能。同時圖3中均未出現cuo的特徵峰(2θ=35.6°、39.8°),說明其合成的分子篩催化劑不存在cuo,或者cuo高度分散的。
實施例2
本實施方式的凝膠製備的加料順序及攪拌時間、水熱反應條件以及後處理過程均按實施例1,不同的是其加料的摩爾比與實施例1不同,本實施方式的原料摩爾比例如表二:
表二
對五組實驗得到的cu-sapo-18分子篩催化劑,分別進行xrf測試,其矽鋁比分別為0.042、0.79、0.11、0.19和0.27。同時說明其分子篩的矽鋁比在較大的範圍內調控。
實施例3
本實施方式的凝膠製備的加料順序及攪拌時間、水熱反應條件以及後處理過程均按實施例1,不同的是其加料的摩爾比與實施例1不同,本實施方式的添加的原料摩爾量比例如表三:
表三
圖5~6是實施例3製備的cu-sapo-18催化劑的催化性能圖,如圖4所示,cu-sapo-18分子篩催化劑在經過三種不同程度的水熱老化條件後,仍具有與新鮮催化劑相似的優異催化活性。圖5表明,鹼土金屬中毒後的催化劑,活性仍然基本得到保持,圖6表明該催化劑晶粒分布均勻。綜上,該分子篩催化劑具有優異的水熱穩定性和抗鹼土金屬中毒性和優良結晶性。