一種越橘提取物的鑑別及其中原花青素含量的測定方法與流程
2023-07-01 05:40:11 5
本發明涉及一種植物提取物的鑑別及其中原花青素含量的測定方法,具體涉及一種越橘提取物的鑑別及其中原花青素含量的測定方法。
背景技術:
原花青素(Proanthocyanidins,PC)是植物中廣泛存在的一大類多酚化合物的總稱,屬於生物類黃酮,由不同數量的兒茶素或表兒茶素聚合而成,其最高平均聚合度達十五聚體。按聚合度的大小,通常將二至四聚體稱為低聚原花青素(簡稱OPC),將五聚體以上的稱為高聚原花青素(簡稱PPC)。原花青素是目前國際上公認的清除人體內自由基最有效的天然抗氧化劑,同時還具有良好的抗癌、抗炎、調節血壓、保護視力、軟化血管等作用,是多種功能性食品的有效成分,其來源廣泛,葡萄籽、銀杏葉、越橘、櫻桃、玫瑰花、松樹皮中均含有原花青素。由於原花青素的多種健康功能,近年來從植物中提取的各種原花青素已廣泛應用於各類保健食品中,其成分鑑別和含量測定是評價保健食品功能的重要指標。
目前對原花青素的檢測方法主要有分光光度法和高效液相色譜法。
分光光度法的基本原理是利用原花青素在無機酸存在和加熱的條件下被降解,產生紅色的花青素,在546nm處有最大吸收,通過分光光度計測定原花青素在水解過程中生成的花青素離子,從而測定原花青素類化合物。此方法具有代表性的是2003版的《保健食品評價與技術規範》。此法雖能測得原花青素總量,但方法中規定的原花青素對照品過於籠統(葡萄籽提取物,純度95%),不易購得,準確度較低,且該方法不能鑑別原花青素成分及來源。
高效液相色譜法的基本原理是樣品經過前處理後,利用HPLC色譜柱將原花青素中的各類組分分離,再用標準品定量。此法不僅可鑑別原花青素的成分及來源,還可測定原花青素含量,但由於原花青素的組分黃烷-3-醇類的立體異構化,原花青素以許多立體異構體形式存在,且各聚合體和同聚異構體之間極性相近,因此,利用HPLC對原花青素進行分離分析和結構表徵非常困難,目前有關利用HPLC對原花青素進行定性定量的研究也僅限於原花青素單體及低聚體。
由於不同來源原花青素提取物的產品價格差異較大,各類原花青素產品檢測標準較低,且檢測主要集中於原花青素含量,導致一些商家將低價格的原花青素產品摻加到高價格產品中,以次充好,擾亂市場,甚至危害食用者的健康和安全。對比市場目前幾個廠家的原料發現,質量不一,液相色譜圖與SW/T 22013國際商務標準中標準品液相色譜圖的出峰情況差別較大,因此市售越橘提取物存在真假混淆的可能。
目前用於鑑別越橘提取物的方法採用SW/T 2-2013國際商務標準經高效液相色譜(HPLC)法進行分析檢測,通過樣品與標準品液相色譜圖的比對鑑別越橘提取物的真假;原花青素含量的測定採用《保健食品檢驗與評價技術規範》2003版「保健食品中原花青素的測定」,通過分光光度法測定原花青素在水解過程中生成的花青素離子,從而測定原花青素類化合物。由于越橘提取物的鑑別和其中原花青素含量的測定方法相互獨立,步驟繁瑣,且通過分光光度法測定原花青素的含量,其測定結果易受越橘提取物中所含花青素的影響,造成測定結果不準確。
技術實現要素:
本發明的目的就是為了克服現有技術中越橘提取物的鑑別和其中原花青素含量的測定方法相互獨立、步驟繁瑣、測定結果不準確的技術問題,提供一種簡單、快速、準確的越橘提取物的鑑別和其中原花青素含量的測定方法。
本發明解決技術問題所採用的技術方案是:採用高效液相色譜法,提供一種越橘提取物的鑑別及其中原花青素含量的測定方法,包括以下步驟:
(1)供試樣品的製備:稱取100mg~150mg供試越橘提取物原料於25mL容量瓶中,加入20mL甲醇,超聲處理20min~30min,冷卻至室溫後,加甲醇定容至刻度,搖勻,以4000r/min離心20~30min後,取上清液備用;
(2)經水解供試品溶液的製備:將正丁醇與鹽酸按95∶5的體積比混合後,製得正丁醇與鹽酸的混合溶液,取出18mL正丁醇與鹽酸的混合溶液置於具塞錐瓶中,再加入0.6mL硫酸亞鐵銨溶液和3mL步驟(1)中製備的試樣溶液,混勻,置98℃~100℃沸水浴回流,加熱1h後立即置2℃~10℃冷水中冷卻,再用10%磷酸水溶液定容至25mL容量瓶中,經0.45μm微孔濾膜過濾後,即得經水解處理的供試品溶液A;
(3)未經水解供試品溶液的製備:取18mL步驟(2)製得的正丁醇與鹽酸混合溶液於25mL容量瓶中,加入0.6mL硫酸亞鐵銨溶液和3mL步驟(1)中製備的試樣溶液,用10%磷酸水溶液定容至刻度,經0.45μm微孔濾膜過濾後,即得未經水解處理的供試品溶液B;
(4)花青素標準品溶液的製備:精確稱取10mg矢車菊素標準品於25mL容量瓶中,加入甲醇定容至刻度,搖勻。從中移取2mL於100mL容量瓶中,用10%磷酸水溶液定容至刻度,經0.45μm微孔濾膜過濾後,即得花青素標準品溶液C;
(5)HPLC分析檢測:精密吸取步驟(2)製得的經水解處理的供試品溶液A、步驟(3)製得的未經水解處理的供試品溶液B、步驟(4)製得的花青素標準品溶液C各15μL,依次注入高效液相色譜儀,得到高效液相色譜圖,其中HPLC色譜條件如下:
a)色譜柱:Agilent C18,4.6×250mm,5μm;
b)流動相A:水∶甲酸=91.5∶8.5,(V/V);流動相B:水∶乙腈∶甲醇∶甲酸=41.5∶22.5∶22.5∶8.5,(V/V/V/V);
梯度條件為0~35min:7~25%B;35~45min:25~65%B;45~46min:65~100%B;46~50min:100%B;50~51min:100~7%B;51~60min:7%B;
c)檢測波長:535nm;
d)流速:1.0mL/min。
(6)越橘提取物真假的鑑別:
將步驟(5)所得的未經水解處理的供試品溶液B的高效液相色譜圖與國際商務標準SW/T 2-2013中的越橘提取物標準品液相色譜圖(圖1所示)進行對比,如供試品為假的越橘提取物,則色譜圖與越橘提取物標準品色譜圖會有明顯不同。
(7)供試樣品中花青素含量的計算:
將步驟(5)所得的經水解處理的供試品溶液A、未經水解處理的供試品溶液B及花青素標準品溶液C的高效液相色譜圖,按外標法分別計算未經水解處理的供試品和經水解處理的供試品中花青素的含量,二者的差值即為試樣中由原花青素水解為花青素的量,從而得出試樣中原花青素的含量。
供試品中游離花青素含量以質量分數w1計,數值以%表示,按下述公式計算:
w1=∑wj
式中:
Wj——供試品中各游離花青素組分的質量分數,%;
Aj——供試品溶液中各游離花青素組分的峰面積(越橘提取物標準品液相色譜圖中峰9,16,18,19,20)
Astd——花青素標準品溶液C中矢車菊素的峰面積;
Cstd——花青素標準品溶液C中矢車菊素的濃度,單位為mg/mL;
m1——供試品的稱樣量,單位為mg;
V1——供試品的定容體積,單位為mL;
P——花青素標準品溶液C中矢車菊素的純度,%;
MWj——供試品溶液中相應的游離花青素組分的分子量;
MWstd——矢車菊素的分子量。
本發明的有益效果:本發明提供一種越橘提取物的鑑別及其中原花青素含量的測定方法,其有益效果在於:
(1)將越橘提取物的鑑別與原花青素含量測定的步驟簡化、結合,供試越橘提取物經簡單的前處理後採用可靠的HPLC分析檢測,即可簡便、準確的在測定越橘提取物中原花青素含量的同時鑑別越橘提取物的真假;
(2)將供試品中的原花青素水解為花青素後,經HPLC檢測花青素含量,將其與未經水解處理的供試品中花青素的含量相減,通過所得差值反映試樣中由原花青素水解為花青素的量,從而得出試樣中原花青素的含量,整個過程中無需考慮試樣中原花青素含有的黃烷醇聚合物的類型和聚合度,同時也可排除試樣中本身含有的花青素對含量測定結果的幹擾;
(3)未經水解處理的供試品溶液的HPLC譜圖,除了用於測定供試品中本身含有的花青素的含量外,還可用於與越橘提取物標準品HPLC譜圖的對比,從而鑑別供試越橘提取物的真假,前處理方法簡單、操作步驟少,分析方法快速、準確,真偽鑑別方法容易、明確。
本發明操作簡便、結果準確,適用於以越橘提取物為原料的保健品、醫藥品和/或化妝品中越橘提取物的鑑別及其中原花青素含量的測定。
附圖說明
圖1為國際商務標準SW/T 2-2013中的越橘提取物標準品HPLC色譜圖;
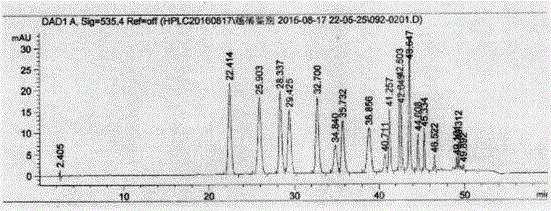
圖2為實施例1中未經水解處理的供試樣品溶液B的HPLC色譜圖;
圖3為實施例1中經水解處理的供試樣品溶液A的HPLC色譜圖;
圖4為實施例2中未經水解處理的供試樣品溶液B的HPLC色譜圖;
圖5為實施例2中經水解處理的供試樣品溶液A的HPLC色譜圖。
具體實施方式
下面結合附圖和具體實施例對本發明作進一步說明,以助於理解本發明的內容。本發明中所使用的方法如無特殊規定,均為常規的生產方法;所使用的試劑,如無特殊規定,均為常規的市售產品。
實施例1
提供一種越橘提取物的鑑別及其中原花青素含量的測定方法,包括以下步驟:
(1)供試樣品的製備:稱取100mg供試越橘提取物原料於25mL容量瓶中,加入20mL甲醇,超聲處理30min,冷卻至室溫後,加甲醇定容至刻度,搖勻,以4000r/min離心20min後,取上清液備用;
(2)經水解供試品溶液的製備:將正丁醇與鹽酸按95∶5的體積比混合後,製得正丁醇與鹽酸的混合溶液,取出18mL正丁醇與鹽酸的混合溶液置於具塞錐瓶中,再加入0.6mL硫酸亞鐵銨溶液和3mL步驟(1)中製備的試樣溶液,混勻,置100℃沸水浴回流,加熱1h後立即置4℃冷水中冷卻,再用10%磷酸水溶液定容至25mL容量瓶中,經0.45μm微孔濾膜過濾後,即得經水解處理的供試品溶液A;
(3)未經水解供試品溶液的製備:取18mL步驟(2)製得的正丁醇與鹽酸混合溶液於25mL容量瓶中,加入0.6mL硫酸亞鐵銨溶液和3mL步驟(1)中製備的試樣溶液,用10%磷酸水溶液定容至刻度,經0.45μm微孔濾膜過濾後,即得未經水解處理的供試品溶液B;
(4)花青素標準品溶液的製備:精確稱取10mg矢車菊素標準品於25mL容量瓶中,加入甲醇定容至刻度,搖勻。從中移取2mL於100mL容量瓶中,用10%磷酸水溶液定容至刻度,經0.45μm微孔濾膜過濾後,即得花青素標準品溶液C;
(5)HPLC分析檢測:用微量移液器吸取步驟(2)製得的經水解處理的供試品溶液A、步驟(3)製得的未經水解處理的供試品溶液B、步驟(4)製得的花青素標準品溶液C各15μL,依次注入高效液相色譜儀,得到高效液相色譜圖,其中HPLC色譜條件如下:
a)色譜柱:Agilent C18,4.6×250mm,5μm;
b)流動相A:水∶甲酸=91.5∶8.5,(V/V);流動相B:水∶乙腈∶甲醇∶甲酸=41.5∶22.5∶22.5∶8.5,(V/V/V/V);
梯度條件為0~35min:7~25%B;35~45min:25~65%B;45~46min:65~100%B;46~50min:100%B;50~51min:100~7%B;51~60min:7%B;
c)檢測波長:535nm;
d)流速:1.0mL/min。
(6)越橘提取物真假的鑑別:
將步驟(5)所得的未經水解處理的供試品溶液B的高效液相色譜圖(圖2所示)與國際商務標準SW/T 2-2013中的越橘提取物標準品液相色譜圖(圖1所示)進行對比,發現圖2與圖1出峰情況相似,判斷該供試品為真的越橘提取物。
因無實際標準品對照,且檢測設備系統適應性不同,會導致出峰時間不同,因此供試樣品與越橘提取物標準品色譜圖的比對主要考慮出峰個數及大致峰面積是否相似。
(7)供試樣品中花青素含量的計算:
將步驟(5)所得的經水解處理的供試品溶液A(圖3所示)、未經水解處理的供試品溶液B及花青素標準品溶液C的高效液相色譜圖,按外標法分別計算未經水解處理的供試品和經水解處理的供試品中花青素的含量,二者的差值即為試樣中由原花青素水解為花青素的量,從而得出試樣中原花青素的含量。
供試品中游離花青素含量以質量分數w1計,數值以%表示,按下述公式計算:
w1=∑wj
式中:
Wj——供試品中各游離花青素組分的質量分數,%;
Aj——供試品溶液中各游離花青素組分的峰面積(越橘提取物標準品液相色譜圖中峰9,16,18,19,20);
Astd——花青素標準品溶液C中矢車菊素的峰面積;
Cstd——花青素標準品溶液C中矢車菊素的濃度,單位為mg/mL;
m1——供試品的稱樣量,單位為mg;
V1——供試品的定容體積,單位為mL;
P——花青素標準品溶液C中矢車菊素的純度,%;
MWj——供試品溶液中相應的游離花青素組分的分子量;
MWstd——矢車菊素的分子量。
經計算,未經水解處理的供試品溶液中游離花青素的含量為4.7%,經水解處理的供試品溶液中游離花青素的含量為53.5%,即試樣中由原花青素水解產生花青素的量為48.8%,從而得出試樣中原花青素的含量為48.8%。
實施例2
提供一種越橘提取物的鑑別及其中原花青素含量的測定方法,包括以下步驟:
(1)供試樣品的製備:稱取150mg供試越橘提取物原料於25mL容量瓶中,加入20mL甲醇,超聲處理35min,冷卻至室溫後,加甲醇定容至刻度,搖勻,以4000r/min離心30min後,取上清液備用;
(2)經水解供試品溶液的製備:將正丁醇與鹽酸按95∶5的體積比混合後,製得正丁醇與鹽酸的混合溶液,取出18mL正丁醇與鹽酸的混合溶液置於具塞錐瓶中,再加入0.6mL硫酸亞鐵銨溶液和3mL步驟(1)中製備的試樣溶液,混勻,置98℃沸水浴回流,加熱1h後立即置8℃冷水中冷卻,再用10%磷酸水溶液定容至25mL容量瓶中,經0.45μm微孔濾膜過濾後,即得經水解處理的供試品溶液A;
(3)未經水解供試品溶液的製備:取18mL步驟(2)製得的正丁醇與鹽酸混合溶液於25mL容量瓶中,加入0.6mL硫酸亞鐵銨溶液和3mL步驟(1)中製備的試樣溶液,用10%磷酸水溶液定容至刻度,經0.45μm微孔濾膜過濾後,即得未經水解處理的供試品溶液B;
(4)花青素標準品溶液的製備:精確稱取10mg矢車菊素標準品於25mL容量瓶中,加入甲醇定容至刻度,搖勻。從中移取2mL於100mL容量瓶中,用10%磷酸水溶液定容至刻度,經0.45μm微孔濾膜過濾後,即得花青素標準品溶液C;
(5)HPLC分析檢測:用微量移液器吸取吸取步驟(2)製得的經水解處理的供試品溶液A、步驟(3)製得的未經水解處理的供試品溶液B、步驟(4)製得的花青素標準品溶液C各15μL,依次注入高效液相色譜儀,得到高效液相色譜圖,其中HPLC色譜條件如下:
a)色譜柱:Agilent C18,4.6×250mm,5μm;
b)流動相A:水∶甲酸=91.5∶8.5,(V/V);流動相B:水∶乙腈∶甲醇∶甲酸=41.5∶22.5∶22.5∶8.5,(V/V/V/V);
梯度條件為0~35min:7~25%B;35~45min:25~65%B;45~46min:65~100%B;46~50min:100%B;50~51min:100~7%B;51~60min:7%B;
c)檢測波長:535nm;
d)流速:1.0mL/min。
(6)越橘提取物真假的鑑別:
將步驟(5)所得的未經水解處理的供試品溶液B的高效液相色譜圖(圖4所示)與國際商務標準SW/T2-2013中的越橘提取物標準品液相色譜圖(圖1所示)進行對比,發現圖4與圖1出峰情況明顯不同,判斷該供試品為假的越橘提取物。
因無實際標準品對照,且檢測設備系統適應性不同,會導致出峰時間不同,因此供試樣品與越橘提取物標準品色譜圖的比對主要考慮出峰個數及大致峰面積是否相似。
(7)供試樣品中花青素含量的計算:
將步驟(5)所得的經水解處理的供試品溶液A(圖5所示)、未經水解處理的供試品溶液B及花青素標準品溶液C的高效液相色譜圖,按外標法分別計算未經水解處理的供試品和經水解處理的供試品中花青素的含量,二者的差值即為試樣中由原花青素水解為花青素的量,從而得出試樣中原花青素的含量。
供試品中游離花青素含量以質量分數w1計,數值以%表示,按下述公式計算:
w1=∑wj
式中:
wj——供試品中各游離花青素組分的質量分數,%;
Aj——供試品溶液中各游離花青素組分的峰面積(越橘提取物標準品液相色譜圖中峰9,16,18,19,20);
Astd——花青素標準品溶液C中矢車菊素的峰面積;
Cstd——花青素標準品溶液C中矢車菊素的濃度,單位為mg/mL;
m1——供試品的稱樣量,單位為mg;
V1——供試品的定容體積,單位為mL;
P——花青素標準品溶液C中矢車菊素的純度,%;
MWj——供試品溶液中相應的游離花青素組分的分子量;
MWstd——矢車菊素的分子量。
經計算,未經水解處理的供試品溶液中游離花青素的含量為2.1%,經水解處理的供試品溶液中游離花青素的含量為17.3%,即試樣中由原花青素水解產生花青素的量為15.2%,從而得出試樣中原花青素的含量為15.2%。
惟以上所述者,僅為本發明的具體實施例而已,當不能以此限定本發明實施的範圍,凡依本發明申請專利範圍所做的均等變化與修飾,皆應屬本發明的涵蓋範圍。