一種製備石墨烯/MOF多孔複合材料水凝膠和氣凝膠的方法與流程
2023-06-10 15:55:36 4
本發明涉及一種石墨烯/金屬有機框架化合物(石墨烯/mof)多孔複合材料水凝膠和氣凝膠的製備方法。
背景技術:
石墨烯(graphene)是一種由碳原子以sp2方式雜化組成的呈六角形蜂巢晶格的單層片狀結構材料(見附圖1)。2004年,geim等成功分離出石墨烯,從而證實它可以單獨在。石墨烯以其獨特的結構使其具有很多優異的電學性能、導熱性及機械強度等獨特的性能,從而被廣泛應用於納米複合材料的製備及電子器件、能量儲存、催化、生物傳感器等領域,顯示出石墨烯的巨大應用潛能。氧化石墨烯是石墨烯的一種溶液分散形式。氧化石墨烯表面和邊緣環氧基團、羥基和羧基基團的存在,不僅能對其進一步改性,而且能提供一個納米範圍的構築結構用以形成新穎的雜化材料,使其在複合材料中具有一定的潛在應用。
金屬-有機框架化合物材料(metal-organicframeworks,mofs)通常是指含氧或氮元素的有機配體與過渡金屬離子通過絡合作用自組裝形成的具有周期性網絡結構的新型納米多孔骨架材料。其中金屬離子可以看作是網絡結構的節點,配體看作連結節點的橋梁。該材料因其巨大的比表面積(~7000m2/g)、特殊的拓撲結構、可控的孔徑尺寸(微孔、介孔)、可調節的功能化及對客體分子的滲透性等特點而在氣體儲存和分離、催化材料、藥物傳遞、成像和傳感、離子交換、光學電子學等領域存在潛在及廣泛的應用前景,引起了科學界的普遍關注。由於mofs材料獨特的框架結構、高孔隙率、化學穩定性、合成工藝簡單以及其潛在的實用價值,其受到了化學工作者的廣泛關注。
近年,基於mofs和石墨烯的納米複合材料因其優異的性能及其在能源、催化、環境、傳感等領域的潛在應用,已經引起了各界人士的廣泛青睞。mofs雖然具有可調的介孔尺寸和較大的比表面積等優點,但是其一般為絕緣或半導體性質。在電化學儲能應用方面,mofs因其相對較低的電子/電荷傳輸速率,在快速的充放電速率下電子轉移受到阻礙,導致較差的倍率性能和循環穩定性。另一方面,石墨烯具有獨特二維原子晶體結構、高理論表面積(2630m2/g)和高電導性,因而是儲能系統中的理想材料。基於mofs和石墨烯兩者的協同作用,在保證兩者結構完整性的基礎上,調控其界面相互作用,通過簡單環保的工藝獲得符合應用需求的石墨烯/mof複合材料已經成為目前該領域的主要研究方向。
目前基於石墨烯/mof複合材料的研究仍然存在著很多問題,尤其是現有合成方法的局限性。石墨烯/mof複合材料的現有合成方法主要包括:溶劑熱或水熱合成法、自組裝法、皮克林乳液法、層層自組裝複合成膜和原子層沉積法等。
方法之一是溶劑熱合成法。該方法是指將金屬鹽、配體、溶劑和石墨烯混合,放入聚四氟乙烯內襯的不鏽鋼反應釜中,密封好後放入烘箱中加熱,隨著反應溫度升高,反應物逐漸溶解,在自生壓力下反應,最終獲得複合材料。優點:一鍋法合成,由於避免了中間操作步驟和提純過程,節約時間和原料成本。缺點:得到產物形貌不可控且石墨烯容易團聚;合成反應溫度高、壓力大,大規模製備具有安全隱患;雖然採用cvd方法可以構築三維石墨烯網絡以避免石墨烯的團聚問題,但其製備條件苛刻,能耗較大。
方法之二是自組裝法。該方法將預先製備的mofs晶體和石墨烯在攪拌混合下獲得石墨烯/mof複合材料粉末。該方法簡單方便,但與上述溶劑熱法具有類似的缺點:(1)石墨烯容易團聚,導致複合物的各組份分布不均勻、導電性差;(2)複合材料大都是粉末狀,沒有較好的微觀形貌和孔道結構;(3)攪拌混合耗時長;(4)石墨烯與mofs之間相互作用力弱,在後處理過程(如洗滌、離心)中mofs晶體易從石墨烯表面脫落,導致最終產物的形貌、組份比例難以控制。雖然目前有報導基於靜電作用將經過表面修飾的帶正電的mofs晶體與氧化石墨烯混合,獲得結構較為穩定的複合材料,但該方法的製備過程較複雜且耗時,表面修飾劑的加入也會影響複合材料的整體性能。
方法之三是皮克林乳液法。該方法是預先製備由有機配體和有機溶劑組成的油相,以及金屬鹽和氧化石墨烯水溶液組成的水相,然後將兩者在適當溫度下攪拌混合,mof會在油/水兩相界面處的石墨烯片上生長,經過純化、洗滌、乾燥得到石墨烯/mof複合材料。該方法由於受到兩相界面的限制,複合材料的形貌難以調節,並且可以合成的mof種類受限,最重要的是難以大規模製備。
方法之四是層層自組裝複合成膜。該方法相對簡單,即將石墨烯溶液(或mof懸浮液)通過真空抽濾(或滴塗)等方法先獲得石墨烯膜(或mof膜),然後在其上面再通過抽濾方法製備一層mof膜(或石墨烯膜)。重複上述步驟,最終獲得「三明治結構」的石墨烯/mof複合膜。該方法雖然簡單,但抽濾工藝耗時長,膜的厚度難以控制,規模化製備仍具有挑戰。
方法之五是原子層沉積法。該方法通過原子層沉積技術在石墨烯片層上先形成金屬氧化物層(如zno或al2o3),再加入有機配體,通過水熱法獲得石墨烯/mof複合材料。該方法雖然簡單,但受到原子層沉積技術可以適用的元素種類局限,可以製備的複合物種類受限。此外,原子層沉積法需要專門的儀器設備,價格昂貴,不易規模化生產。
綜上所述,如何在一般條件下,利用普通的設備儀器,在相對溫和的條件下,獲得形貌和結構可控,組份分布均勻的多孔複合材料,並在此基礎上獲得各種優異性能的多孔複合材料,是該應用領域亟待解決的關鍵技術問題。
技術實現要素:
本發明的目的是提供一種利用簡單混合工藝,基於「溶膠-凝膠」原理,製備形貌和結構可控、組分分布均勻的石墨烯/mof多孔複合材料水凝膠和氣凝膠的方法,該製備方法條件溫和、操作簡單、所用試劑儀器來源廣泛,製得的多孔複合材料氣凝膠既具有三維框架結構且具有多種可調孔徑,能保留石墨烯和mofs結構的完整性,並能夠有效阻止石墨烯片或氧化石墨烯片以及mofs晶體的團聚。
為實現上述發明目的,本發明採用如下技術方案:
一種石墨烯/mof多孔複合材料水凝膠和氣凝膠的製備方法,所述製備方法為:取一乾淨容器,向其中加入石墨烯或氧化石墨烯分散液,然後向其中加入mofs晶體粉末,使mofs晶體粉末與石墨烯或氧化石墨烯的投料質量比為1:100~100:1,密封容器後進行震蕩或攪拌,促使石墨烯或氧化石墨烯自組裝三維框架結構的形成,以及mofs晶體與石墨烯片或氧化石墨烯片均勻複合,從而獲得石墨烯/mof多孔複合材料水凝膠,最後經冷凍乾燥獲得石墨烯/mof多孔複合材料氣凝膠,所述石墨烯/mof多孔複合材料氣凝膠具有自支撐的多孔結構,保留了石墨烯或氧化石墨烯和mofs結構的完整性。
進一步,mofs晶體粉末與石墨烯或氧化石墨烯的投料質量比優選為1:1~10:1。
進一步,mofs晶體與石墨烯片或氧化石墨烯片表面的複合方式包括但不限於:mofs晶體在石墨烯片或氧化石墨烯片表面的均勻附著,mofs晶體被石墨烯片或氧化石墨烯片包覆等。在一種石墨烯/mof多孔複合材料水凝膠或氣凝膠中,上述複合方式可以單一存在,也可以組合存在,具體視mofs晶體本身性質和加入量而定。
本發明所採用的氧化石墨烯分散液中,氧化石墨烯依靠本身π-π堆疊和範德華吸引力與其表面含氧官能團間的排斥力達到平衡穩定的分散。本發明採用溶膠-凝膠法,向穩定分散的氧化石墨烯溶液中加入mofs晶體,打破了這種平衡,導致氧化石墨烯有序堆砌形成水凝膠,經冷凍乾燥獲得氣凝膠。因此,從本發明的成膠機理分析,無論是以何種方法獲得的氧化石墨烯分散液,只要加入mofs晶體能夠打破其平衡狀態,都能獲得凝膠。同理,無論是何種方法(即使是依靠某些輔助方法例如表面活性劑等)獲得的穩定分散的石墨烯分散液,只要加入mofs晶體能夠打破其平衡狀態,也能獲得凝膠。本發明優選所述石墨烯或氧化石墨烯分散液中,石墨烯或氧化石墨烯的橫向尺寸控制在0.1μm~100μm範圍內,更優選在1~10μm之間;石墨烯或氧化石墨烯的濃度控制在0.1~100mg/ml範圍之間,更優選為1~10mg/ml。
本發明具體推薦了通過改性hummer法製備氧化石墨烯,具體方法如下:在冰水浴條件下,將石墨粉、硝酸鹽按照質量比1:(0.01~100)加入質量濃度為95%-98%的濃硫酸中混合均勻,向上述混合溶液中加入高錳酸鉀,其中高錳酸鉀與石墨粉的投料質量比為(0.1~10):1,並控制溫度不超過10℃下保持0.5~10h(優選2h);然後去掉冰水浴,加熱到10~100℃(優選35℃)並保持10~120分鐘(優選30分鐘);向其中加入水,攪拌,再向其中加入雙氧水還原剩餘的高錳酸鉀和mno2;離心並用熱水清洗殘渣直至上述懸浮液的ph為7;把得到的粉末再次分散到水中超聲,過濾後得到懸浮液即氧化石墨烯分散液。硝酸鹽優選自下列化學純和分析純之一:硝酸鉀,硝酸鈉,更優選硝酸鈉。
本發明幾乎適用於所有mofs晶體,在製備過程中,可加入一種mofs,也可以加入兩種以上mofs。mofs晶體可通過文獻報導的方法進行製備,例如ni-mof、fe-mof、zif-8、mof-5、co-mof和[k2sn2(bdc)3](h2o)x晶體,均可通過溶劑熱法合成。具體而言,溶劑熱法可按照如下進行:將金屬鹽或金屬鹽水合物、有機配體及溶劑按比例進行混合,然後對混合物進行溶劑熱反應獲得mofs晶體沉澱,進一步通過離心或靜置處理、真空乾燥獲得mofs晶體粉末。所述的金屬鹽或金屬鹽水合物中金屬中心的選擇幾乎覆蓋了所有金屬,包括主族元素、過渡元素、鑭系金屬等,其中應用較多的為zn、cu、fe、ni、co、sn等。有機配體可選自下列化學純或分析純藥品之一:羧酸類、咪唑類、吡啶類、卟啉類等。溶劑選自下列化學純或分析純試劑之一:甲醇、乙醇、n,n-二甲基甲醯胺、去離子水等。
本發明所製備的石墨烯/mof多孔複合材料具有自支撐的多孔結構,在合成過程中保留了石墨烯和mofs結構的完整性,兼具石墨烯和mofs晶體的優異性能,有望在儲能、催化、吸附、傳感等領域中同時發揮石墨烯和mofs兩者的優異性能,具有潛在的應用前景。
本發明相對於現有技術具有如下突出的優點和有益效果:
第一,本發明藉助石墨烯或氧化石墨烯溶液的性質,基於「溶膠-凝膠」原理,通過加入不同種類的mofs晶體,促使石墨烯或氧化石墨烯自組裝三維框架結構的形成,以及mof晶體在石墨烯片或氧化石墨烯片表面的均勻附著。通過本發明獲得的石墨烯/mof複合材料氣凝膠,具有自支撐的多孔結構,保留了石墨烯和mofs結構的完整性,兼具石墨烯或氧化石墨烯和mofs晶體的優異性能。此外,本發明可以同時引入兩種以上的mofs,從而獲得含有多種mofs的石墨烯/mof多孔複合材料氣凝膠。
第二,本發明具有普適性,幾乎適用於所有mofs。此外,本發明對於石墨烯和氧化石墨烯也無特殊要求,只要是穩定分散的石墨烯分散液或氧化石墨烯分散液均適用於本發明。因此基於本發明可製備具有各種不同功能特性的石墨烯/mof多孔複合材料氣凝膠,以適用於不同的應用領域。
第三,本發明便於批量化或工業化生產。本發明採用一步法,僅僅通過攪拌混合mof與石墨烯或氧化石墨烯兩種材料即可實現;用到的溶劑在實驗室或工業化生產中來源廣泛、價格低廉,試驗設備儀器簡單、操作方便,並且在合成過程中保留了石墨烯或氧化石墨烯和mofs結構的完整性,並能夠有效阻止石墨烯片或氧化石墨烯片以及mofs晶體的團聚。
附圖說明
圖1:石墨烯結構示意圖。
圖2:純石墨烯氣凝膠(a,b)、fe-mof(c,d)和ni-mof(e,f)晶體掃描電鏡圖。
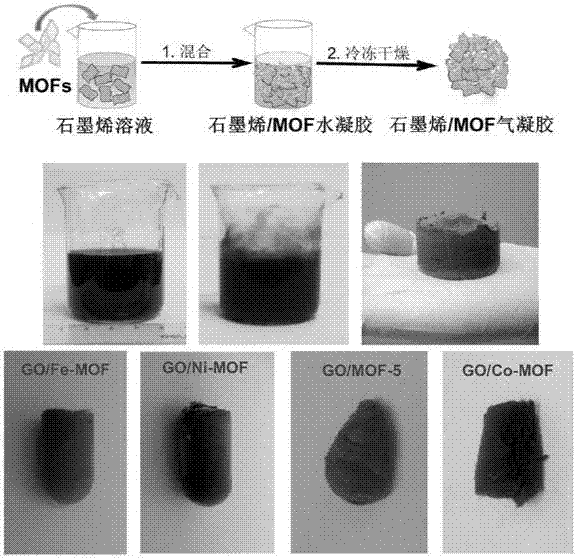
圖3:石墨烯/mof複合材料氣凝膠製備工藝流程圖及石墨烯水相分散液、各種石墨烯/mof複合材料水凝膠和氣凝膠的照片。
圖4:不同質量比的石墨烯/fe-mof複合材料氣凝膠的掃描電鏡圖:fe-mof:go=1:1(a,b);fe-mof:go=2:1(c,d);fe-mof:go=4:1(e,f)。
圖5:石墨烯/fe-mof複合材料氣凝膠的掃描電鏡圖(fe-mof:go=5:1(a,b))及其透射電鏡圖(c,d)。
圖6,圖7:分別是圖5所示的石墨烯/fe-mof複合材料氣凝膠的xrd,raman及ftir圖,輔助圖5中掃描電鏡圖證明石墨烯與mof均勻複合。
圖8:不同質量比的石墨烯/ni-mof複合材料氣凝膠的掃描電鏡圖:ni-mof:go=1:1(a,b);ni-mof:go=3:1(c,d)。
圖9:石墨烯/ni-mof複合材料氣凝膠的掃描電鏡圖(ni-mof:go=5:1(a,b))及其透射電鏡圖(c,d)。
圖10,圖11:分別是圖9所示石墨烯/ni-mof複合材料氣凝膠的xrd,raman及ftir圖。
圖12:不同質量比的石墨烯/fe-mof/ni-mof複合材料氣凝膠的掃描電鏡圖:(fe-mof&ni-mof):go=(5+1):1(a,b);(fe-mof&ni-mof):go=(5+3):1(c,d);(fe-mof&ni-mof):go=(5+5):1(e,f)。
圖13:純zif-8晶體和不同質量比的石墨烯/zif-8複合材料氣凝膠的掃描電鏡圖:zif-8晶體(a,b);zif-8:go=3:1(c,d);zif-8:go=5:1(e,f)。
圖14:純mof-5晶體和石墨烯/mof-5複合材料氣凝膠的掃描電鏡圖:mof-5晶體(a,b);mof-5:go=5:1(c,d);mof-5:go=10:1(e,f)。
圖15:純[k2sn2(bdc)3](h2o)x晶體和石墨烯/[k2sn2(bdc)3](h2o)x複合材料氣凝膠的掃描電鏡圖:[k2sn2(bdc)3](h2o)x晶體(a);[k2sn2(bdc)3](h2o)x:go=5:1(b,c)。
圖16:純co-mof晶體和石墨烯/co-mof複合材料氣凝膠的掃描電鏡圖:co-mof晶體(a);co-mof:go=10:1(b,c)。
具體實施方式:
下面結合具體實施例和附圖對本發明作進一步詳細的描述,但本發明的實施方式並不僅限於此。
實施例1~6
1、樣品的製備
(1)實施例1樣品的製備按如下步驟進行:
在冰浴中配置好250ml的反應瓶,加入96ml的濃硫酸,磁力攪拌下加入2g石墨粉和1g硝酸鈉(>99%)固體混合物,再緩慢加入6g高錳酸鉀(>99.6%),控制反應溫度不超過10℃,在冰浴條件下反應2h後取出,在水浴35℃下攪拌18h。隨著反應的進行,反應物最終變成棕色漿狀,接著向其中緩慢加入質量分數為5%的硫酸溶液進行稀釋。加入240ml硫酸溶液後,向其中加入5ml質量分數為30%的雙氧水,隨著氣泡的鼓出,溶液變成亮黃色。持續攪拌2h後過濾,然後用質量分數10%的鹽酸溶液、去離子水多次洗滌,直至溶液呈中性。最終將獲得的產物以濃度為10mg/ml的go水相分散液的形式保存備用。
(2)實施例2樣品的製備按如下步驟進行:
在室溫下配置好100ml的反應瓶,加入50ml的n,n-二甲基甲醯胺,磁力攪拌下加入1.215g的無水氯化鐵和0.83g對苯二甲酸,完全溶解後,在油浴100℃下反應12h。反應結束後,低速離心(室溫,4000rpm,15min),去除上清液,然後用n,n-二甲基甲醯胺溶劑重複洗滌、離心3次。所得產物經60℃真空乾燥24h後最終獲得fe-mof晶體粉末。
(2)實施例3樣品的製備按如下步驟進行:
在5ml大小的離心管中依次加入氧化石墨烯水相分散液、fe-mof晶體粉末,控制各原料的初始投料比如下:氧化石墨烯溶液2ml,濃度4mg/ml;fe-mof晶體粉末8mg(mgo﹕mfe-mof=1∶1);所得混合物在q2-1型漩渦混合器上持續混合2min,獲得go/fe-mof複合材料水凝膠。進一步對其進行冷凍乾燥24h,最終獲得go/fe-mof複合材料氣凝膠。
(3)實施例4~6樣品的製備
實施例4~6樣品的製備步驟同實施例3,不同之處在於,實施例4~6中fe-mof晶體粉末的質量分別為16mg(mgo﹕mfe-mof=1∶2),32mg(mgo﹕mfe-mof=1∶4),40mg(mgo﹕mfe-mof=1∶5)。
2、表徵與測試
(1)sem分析
sem測試在hitachis-4700掃描電子顯微鏡上進行,所用樣品製備方法如下:取上述所得4mg/mlgo水相分散液2ml通過液氮迅速冷凍後,放入冷凍乾燥機中-56℃下冷凍乾燥24h後獲得,取少量置於貼有導電膠的支持臺表面。同樣,取少量上述獲得的fe-mof晶體粉末置於同一支持臺上不同位置,隨後將其放入sem腔室中進行測試。實施例3~6所獲得的不同比例的go/fe-mof複合材料氣凝膠,與實施例1相同。
(2)高解析度tem分析
tem測試在jeol2010f型透射電子顯微鏡上進行,所用樣品製備方法如下:取上述實施例6中go/fe-mof複合材料氣凝膠樣品微量,置於裝有1ml去離子水的小瓶中,然後將其放入超聲功率為250w的水浴超聲池內於25℃恆溫下被持續超聲0.5h,獲得複合材料水相分散液,取少量分散液滴在tem帶微柵銅網表面(含微孔碳支持膜),隨後室溫下自然乾燥獲得。
(3)廣角xrd分析
xrd測試在x'pertpro型x射線衍射儀上進行,待測樣品製備如下:取實施例2中fe-mof晶體粉末,鋪於石英片上方形磨砂凹槽中,並用載玻片壓實壓平經行測試。實施例6,與實施例2相同。
(4)raman光譜分析
raman測試在labramhr800型拉曼光譜儀上進行,樣品製備如下:取實施例1中純go氣凝膠少量,置於乾淨載玻片上壓平製得。實施例6,同實施例1。
(5)ftir光譜分析
ftir測試在nicolet6700傅立葉變換紅外光譜儀上進行,在這裡我們採用ar法測試,樣品直接用實施例1、2和實施例6獲得的試樣。
3、測試結果的比較與分析
實施例1中所獲得的純go氣凝膠的表面形貌如圖2(a,b)。圖2(a)展示了氣凝膠的整體形貌,可以看出三維框架是由大量go片搭接而成,並構造出無數微米級孔道。圖2(b)是圖2(a)局部放大圖,從(b)可以明顯看出go片是透明的,說明go片沒有發生團聚,進一步證明這一自支撐的多孔結構是由大量單層go片構築而成。圖2(c,d)是實施例2中獲得的fe-mof晶體粉末的sem圖,從圖(d)可以看出,獲得的fe-mof晶體是多邊形紡錘體,橫向尺寸為幾百個納米。
圖4和圖5(a,b)分別是實施例3~6中獲得的go/fe-mof複合材料氣凝膠的sem圖。低倍率的掃描電鏡圖可以看出,他們都具有三維網絡結構和微米級孔道,並未太大區別。從高倍率掃描電鏡圖4(b,d,f)和圖5(b)則能明顯看出,fe-mof晶體均勻的附著在go片表面,與實施例2中fe-mof晶體相比,形貌沒有發生變化。不同之處在於,隨著fe-mof晶體加入量的增多,go片上附著的fe-mof晶體隨之增加。圖5(b)中fe-mof晶體已經將go片表面全部鋪滿,並且沒有發現fe-mof團聚現象,直觀的證明了fe-mof晶體和go片被均勻的複合,並成功獲得三維框架結構。
圖5(c,d)展示了實施例6樣品的tem照片,圖中顯示fe-mof晶體均勻附著在go片表面,與實施例1(圖2(b))相比,go片更加褶皺,這可能是fe-mof晶體附著其表面所致;與實施例2(圖2(d))相比,fe-mof晶體形貌和尺寸沒有變化。這表明go和fe-mof複合過程中保留自身的完整結構。
圖6(a)給出了實施例2和實施例6中所制兩樣品的廣角xrd譜圖,圖中顯示在實施例2中樣品存在的峰,在實施例6樣品中都能找到對應的衍射峰,說明實施例6中獲得的複合材料氣凝膠中包含實施例2中fe-mof晶體,並且晶體結構保持完整。不同之處,實施例6樣品xrd譜圖在約26°處存在一個大的峰包,而這個峰包是石墨烯(002)晶面特徵衍射峰,證明石墨烯結構的存在(即go)。xrd譜圖分析表明,go與fe-mof被成功複合,這與上述sem和tem分析結果相同。
圖6(b)進一步比較了實施例1和實施例6所對應的raman譜圖。在實施例1所對應的樣品即go氣凝膠的譜圖和實施例6所對應的樣品即go/fe-mof複合材料氣凝膠的譜圖中,d峰和g峰的位置有所偏移,特別是d峰的位置,說明go和fe-mof兩者之間有相互作用力,主要是兩者之間的靜電相互作用。我們知道,id/ig對應於石墨烯中缺陷、sp3雜化與sp2雜化鍵的相互關係。實施例6中id/ig強度與實施例1相比明顯增大,表明go與fe-mof相互作用後,形成了更多石墨化碳和sp3雜化鍵。
圖7給出了實施例1,2和實施例6所對應的ftir譜圖。實施例2譜圖中顯示,1383cm-1和1498cm-1處有兩個明顯的吸收峰,這對應與對苯二甲酸中羧酸基團的對稱伸縮振動峰;1591cm-1處強烈的吸收峰,歸功於c-c和吸收的-oh基團的伸縮共振峰。實施例2和實施例6的ftir譜圖對比,發現兩者幾乎重合,說明實施例6中樣品包含實施例2中樣品。但是實施例6譜圖中沒有發現實施例1樣品譜圖的吸收峰,這是實施例6中樣品中實施例1樣品含量太低(16.7wt%)導致。綜上所述,通過我們的方法可以成功製備各組分分布均勻、形貌結構可控的go/fe-mof複合材料氣凝膠。
實施例7~10
1、樣品的製備
(1)實施例7樣品的製備按如下步驟進行:
第1步:在50ml大小的燒杯中加入20ml去離子水,然後依次向其中加入1.3g六水合氯化鎳和3g草酸鈉,攪拌使其完全溶解,此時溶液呈綠色。
第2步:另取一個50m大小的燒杯,向其中加入1.3g六水合氯化鎳,使其平鋪於燒杯底部,然後緩慢滴加1.5ml的乙二胺溶液,使其與六水合氯化鎳充分接觸並反應,此時樣品呈深紫色。需要注意:滴加乙二胺的過程會放出大量熱,所以滴加過程儘量慢且不能攪拌。
第3步:將上述步驟2中樣品加入步驟1中的燒杯中,磁力攪拌使其完全溶解,然後室溫條件下持續攪拌反應48h。反應結束後,低速離心(室溫,4000rpm,15min),去除上清液,然後用甲醇重複洗滌、離心3次.所得產物經60℃真空乾燥24h後最終獲得ni-mof晶體粉末。
(2)實施例8樣品的製備按如下步驟進行:
在5ml大小的離心管中依次加入氧化石墨烯水相分散液、ni-mof晶體粉末,控制各原料的初始投料比如下:氧化石墨烯溶液2ml,濃度4mg/ml;ni-mof晶體粉末8mg(mgo﹕mni-mof=1∶1);所得混合物在q2-1型漩渦混合器上持續混合2min,獲得go/ni-mof複合材料水凝膠。進一步對其進行冷凍乾燥24h,最終獲得go/ni-mof複合材料氣凝膠。
(3)實施例9,10樣品的製備:
實施例9,10樣品的製備步驟同實施例7,不同之處在於,實施例9,10中ni-mof晶體粉末的質量分別為24mg(mgo﹕mni-mof=1∶3),40mg(mgo﹕mni-mof=1∶5)。
2、表徵測試
(1)sem分析
與實施例1~6相同。
(2)高分辨tem分析
對實施例10進行高分辨tem測試,與實施例6相同。
(3)廣角xrd分析
對實施例7和實施例10進行xrd測試,與實施例2、實施例6相同。
(4)raman光譜分析
對實施例10進行raman測試,與實施例1,2、實施例6相同。
(5)ftir光譜分析
對實施例7和實施例10進行raman測試,與實施例1,2、實施例6相同。
3、對測試結果的比較與分析
實施例7中所獲得的ni-mof晶體表面形貌如圖2(e,f)。圖2(e)展示了低倍率下ni-mof形貌,圖2(f)是圖2(e)局部放大圖,可以看出ni-mof是直徑約500納米,長5~10微米的棒狀晶體。
圖8和圖9(a,b)分別是實施例8~10中獲得的go/ni-mof複合材料氣凝膠的sem圖。同實施例3~6,我們分別觀察了其低倍率和高倍率下的表面形貌。同樣低倍率的掃描電鏡圖(圖8(a,c)和圖9(a))可以看出,他們都具有三維網絡結構和微米級孔道。從高倍率掃描電鏡圖8(b,d)和圖5(b)則能明顯看出,ni-mof晶體均勻的附著在go片表面,與實施例7中ni-mof晶體相比,形貌沒有發生變化。與實施例3~6相同,隨著ni-mof晶體加入量的增多,go片上附著的ni-mof晶體隨之增加。圖9(b)中go片表面全部鋪滿ni-mof晶體,並且沒有發現ni-mof團聚現象,同樣,證明了ni-mof晶體和go片被均勻的複合,並成功獲得三維框架結構。
圖9(c)給出了實施例10中樣品的tem圖。圖中給出了實施例10樣品局部放大圖,顯示ni-mof晶體表面被go片包裹。結合圖9(b)我們得出,實施例10樣品中ni-mof晶體部分被go片包覆,部分附著在go片表面,共同構成go/ni-mof複合材料氣凝膠。
圖10(a)比較了實施例7和實施例10樣品的xrd圖。同樣,實施例7樣品中所具有的特徵衍射峰,在實施例10樣品的xrd圖中都能找到對應的衍射峰,說明實施例10樣品中包含實施例7樣品。實施例10樣品相對實施例7樣品的xrd圖,在2θ≈26°處出現一個明顯的峰包,而這是石墨烯(002)晶面特徵衍射峰,表明實施例10樣品中同時含有go片和ni-mof晶體,進一步證明了go片和ni-mof晶體成功複合,並且ni-mof晶體保持原有完整結構。
圖10(b)比較了實施例1和實施例10樣品的raman圖。可以看出,實施例10樣品的raman光譜圖中d峰和g峰的位置相對於實施例1樣品d峰和g峰的位置發生了偏移,表明ni-mof晶體加入後與go片發生了相互作用,這種作用歸功於ni-mof與go之間的靜電相互作用。同比較例4,ni-mof晶體加入後,id/ig明顯增加,說明更多石墨化碳和sp3雜化鍵形成,同時也佐證了ni-mof晶體與go片之間具有相互作用。
圖11比較了實施例1,實施例7和實施例10樣品的ftir光譜圖。實施例10樣品的特徵吸收峰與實施例7樣品的吸收峰幾乎重合,說明實施例10樣品中含有實施例7樣品。同樣,在實施例10樣品的ftir吸收峰中沒有發現實施例1樣品的特徵吸收峰,主要因為實施例1樣品在實施例10樣品中含量太低(16.7wt%)。綜上可知,通過我們的方法可以成功製備各組分分布均勻、形貌結構可控的go/ni-mof複合材料氣凝膠。
實施例11~13
1、樣品的製備
(1)實施例11樣品的製備按如下步驟進行:
在5ml大小的離心管中依次加入氧化石墨烯水相分散液、fe-mof和ni-mof晶體粉末,控制各原料的初始投料比如下:氧化石墨烯溶液2ml,濃度4mg/ml;fe-mof晶體粉末40mg,ni-mof晶體粉末8mg(mgo﹕m(fe-mof&ni-mof)=1∶(5+1));所得混合物在q2-1型漩渦混合器上持續混合2min,獲得go/fe-mof&ni-mof複合材料水凝膠。進一步對其進行冷凍乾燥24h,最終獲得go/fe-mof&ni-mof複合材料氣凝膠。
(2)實施例12,13樣品的製備
實施例12,13樣品的製備步驟同實施例11樣品的製備,不同之處在於,實施例12樣品製備過程中加入ni-mof晶體粉末的質量為24mg(mgo﹕m(fe-mof&ni-mof)=1∶(5+3)),實施例13樣品中ni-mof晶體的質量為40mg(mgo﹕m(fe-mof&ni-mof)=1∶(5+5))。
2、表徵測試
(1)sem分析
與實施例1,實施例8-10相同。
3、對測試結果的比較與分析
圖12(a,c,e)分別給出了實施例11-13樣品低倍率的sem圖,可以看出實施例11-13樣品均是具有微米級孔道的三維框架結構。圖12(b,d,f)分別是圖12(a,c,e)中對應的局部放大圖,如圖12(b)所,go片包覆在ni-mof晶體棒表面,fe-mof晶體附著在go片表面,最終構成了go/fe-mof&ni-mof複合材料氣凝膠。綜上可知,採用我們的方法不僅可以製備出go和單種mof複合材料氣凝膠,同時可以獲得go和兩種以上mof複合材料氣凝膠。
實施例14-16
1、樣品的製備
(1)實施例14樣品的製備按如下步驟進行:
第1步:在50ml大小的燒杯中加入15ml甲醇,然後向其中加入18.9g六水合硝酸鋅,攪拌使其完全溶解。
第2步:另取一個50m大小的燒杯中加入30ml甲醇,向其中加入8.2g的2-甲基咪唑,攪拌使其完全溶解。
第3步:將上述步驟2中樣品加入步驟1中的燒杯中,磁力攪拌使其混合均勻,然後室溫條件下持續攪拌反應12h。反應結束後,低速離心(室溫,4000rpm,15min),去除上清液,然後用甲醇重複洗滌、離心3次.所得產物經60℃真空乾燥24h後最終獲得zif-8晶體粉末。
(2)實施例15,16樣品的製備
實施例15,16樣品的製備同實施例3樣品製備。不同之處,實施例3中加入實施例2樣品,實施例15,16中加入實施例14樣品。實施例15樣品製備中,加入實施例14樣品24mg(mgo:mzif-8=3:1);實施例16樣品製備同實施例15樣品製備,不同在於實施例16中加入實施例14的質量為40mg(mgo:mzif-8=5:1)。
2、表徵測試
(1)sem分析
實施例14-16,同實施例4,實施例1-2,7,14。
3、對測試結果的比較與分析
圖13(a,b)給出了實施例14樣品的sem圖。從圖13(b)可以看出,zif-8晶體呈微米級別片狀,純zif-8晶體粉末是大量zif-8晶體片堆砌而成。
圖13(c,d)和(e,f)分別是實施例15,16樣品的sem圖。圖13(c)和(e)是實施例15,16樣品低倍率sem圖,從圖可知,實施例15,16樣品是具有微米孔道的氣凝膠。圖13(d)和(f)分別是圖13(c)和(e)樣品局部放大圖。以實施例16為例,從圖13(f)可以看出,相比實施例1樣品的sem圖2(b),實施例16樣品中zif-8晶體片附著在go片表面,導致go片不再像實施例1樣品那樣透明。同時,與實施例14樣品相比,實施例16樣品中zif-8晶體沒有發生團聚。同樣,通過我們的方法可以獲得分組分分布均勻,形貌結構可控的go/zif-8複合材料氣凝膠。
實施例17~19
1、樣品製備
(1)實施例17樣品製備步驟如下:
在50ml大小的燒杯中加入12mln,n-二甲基甲醯胺,然後依次向其中加入0.357g的六水合硝酸鋅,0.066g的對苯二甲酸,室溫條件下攪拌使其完全溶解並混合均勻,然後將其轉移到反應釜中密封,在烘箱中120℃反應24h。反應結束後,低速離心(室溫,4000rpm,15min),去除上清液,然後用dmf重複洗滌、離心3次.所得產物經60℃真空乾燥24h後最終獲得mof-5晶體粉末。
(2)實施例18,19樣品製備:
實施例18,19樣品製備同實施例3樣品製備。不同的是,實施例18,19樣品中加入的晶體粉末是mof-5,實施例18,19樣品中加入mof-5質量分別為40mg((mgo:mmof-5)=5:1)和80mg((mgo:mmof-5=10:1))。
2、表徵測試
(1)sem分析
實施例17-19,與實施例2、實施例3相同。
3、對測試結果的比較和分析
圖14(a,b)展示了實施例17樣品的sem圖。同樣,我們對實施例17樣品分別進行了低倍率(圖14(a))和高倍率(圖14(b))下sem測試。從圖14(b)可知,實施例17獲得的樣品呈無規多邊形,尺寸為幾個微米。
圖14(c,d)和圖14(e,f)分別是實施例18,19樣品的sem圖。圖14(c)和(e)展示了實施例18,19樣品的整體形貌,證明了實施例18,19樣品具有三維框架結構。圖14(d)和(f)分別是(c)、(f)局部放大圖,從中可以看出,隨著mof-5晶體量的增加,mof-5晶體將go片表面全部覆蓋,go片也不再如實施例1樣品那樣透明,表明go與mof-5已成功複合,並且複合過程可以同時防止go和mof-5發生團聚。綜上可知,通過我們的方法也可以獲得各組分分布均勻,結構形貌可控的go/mof-5複合材料氣凝膠。
實施例20-21
1、樣品製備
(1)實施例20樣品製備按如下步驟進行:
在50ml大小的燒杯中加入10ml去離子水,然後依次向其中加入0.128g的硫酸亞錫,0.099g的對苯二甲酸,56mg氫氧化鉀,室溫下混合攪拌使其完全溶解,然後將其轉移到反應釜中180℃反應15h。反應結束後,低速離心(室溫,4000rpm,15min),去除上清液,然後用去離子水重複洗滌、離心3次.所得產物經60℃真空乾燥24h後最終獲得[k2sn2(bdc)3](h2o)x晶體粉末。
(2)實施例21樣品製備:
同實施例3,不同的是,實施例3樣品加入實施例2樣品,實施例21樣品加入實施例20樣品,加入量為40mg((mgo:m[k2sn2(bdc)3](h2o)x)=5:1)。
2、表徵測試
(1)sem分析
實施例20-21,與實施例2、實施例3相同。
3、對測試結果的分析與比較
圖15(a)展示了實施例20樣品的sem圖。從圖可以看出,[k2sn2(bdc)3](h2o)x晶體呈微米級短棒狀。
圖15(b,c)分別是實施例21樣品的sem圖。圖(b)表明實施例21是三維框架結構,圖(c)表明實施例20成功附著在go片上(與實施例1比較),證明通過我們的方法成功獲得了go/[k2sn2(bdc)3](h2o)x複合材料氣凝膠。
實施例22-23
1、樣品製備
(1)實施例22樣品製備過程如下:
第1步,在50ml大小的燒杯中加入20ml甲醇和20ml乙醇的混合溶液,向其中加入725mg的六水合硝酸鈷,攪拌使其完全溶解。
第2步,在另一50ml大小的燒杯中加入20ml甲醇和20ml乙醇的混合溶液,向其中加入821mg的2-甲基咪唑,攪拌使其完全溶解。
第3步,將上述兩燒杯中的溶液混合攪拌均勻,室溫下反應24h。反應結束後,速離心(室溫,4000rpm,15min),去除上清液,然後用甲醇重複洗滌、離心3次,所得產物經60℃真空乾燥24h後最終獲得co-mof晶體粉末。
(2)實施例23樣品製備:
實施例23樣品製備,同實施例3。不同之處,實施例23樣品加入的是實施例22樣品,加入量為80mg(mgo:mco-mof=10:1)。
3、對測試結果的分析與比較
圖16(a)給出了實施例22樣品的sem圖。圖中展示了實施例23樣品是尺寸為幾百納米大小的小顆粒。
圖16(b,c)展示了實施例23樣品的表面形貌。圖16(b)是低倍率sem圖,從圖可知,實施例23樣品是由實施例1和實施例23樣品共同構築的三維框架結構,並且三維框架結構具有微米級孔道。圖16(c)是圖16(b)樣品的局部放大圖,圖中展示了實施例22樣品均勻的附著在實施例1樣品表面,並將實施例1樣品完全包覆,同時實施例1樣品還保持原有片狀,表明通過我們的工藝步驟也能獲得go/co-mof複合材料氣凝膠。