金屬空氣電池用電極的製作方法
2023-06-09 01:19:46 1
本發明涉及一種將水溶液用作電解液的金屬空氣電池用電極,特別涉及一種以氧為活性物質的正極的催化劑層。
背景技術:
一般地說,將水溶液用作電解液的金屬空氣電池是組合有負極反應即金屬的氧化還原反應、和正極反應即氧的還原反應與生成反應的電池,它可以從空氣中吸取作為正極活性物質的氧,從而在正極可以省略保持活性物質的空間,因而可以期待成為具有高能量密度的電池。
例如在負極金屬為鋅的情況下,兩極的放電反應如以下那樣進行。
正極:o2+2h2o+4e-→4oh-
負極:2zn+4oh-→2zno+2h2o+4e-
此外,逆反應為充電反應,金屬空氣電池無論是作為一次電池還是作為二次電池都可以發揮作用。
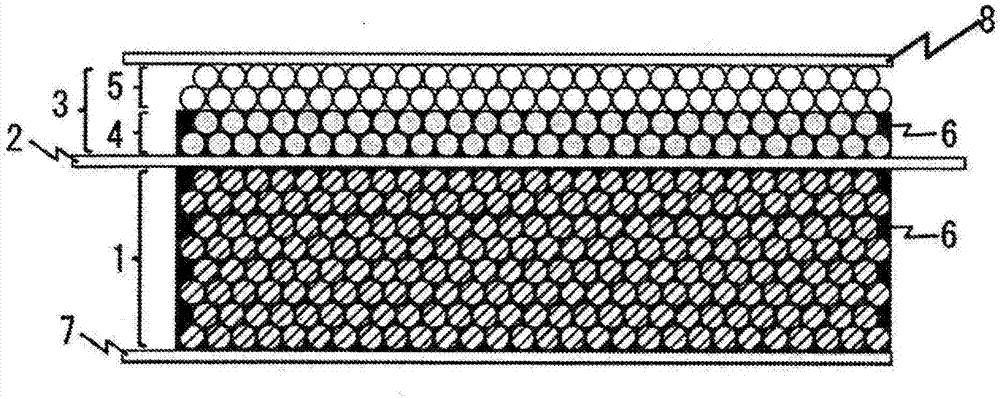
金屬空氣電池的通常的結構如圖1以及圖2所示。金屬空氣電池具有負極1、隔膜(或者電解質膜)2、正極3、催化劑層4、多孔質層5、電解質水溶液6、負極集電體7、正極集電體8。金屬空氣電池形成負極1和正極3以不會產生物理接觸的方式而被隔膜2分隔的結構。隔膜2形成為如下的結構:為了使強鹼性的電解質水溶液6能夠透過而採用多孔質材料、或者可以傳導oh-離子的高分子膜形成,電解質水溶液6滲入負極1和正極3的多孔質空隙,充放電時oh-可以經由電解質水溶液而在兩極間移動。特別地,正極3的材質為了確保電子傳導性而大多使用具有電子傳導性的碳材料作為主成分,其外側(隔膜的相反側)向大氣開放,或者曝露於供給空氣的流路中,從而正極放電反應所需要的空氣可以向正極內擴散。
再者,為了降低正極反應的過電位,在正極內配置有催化劑。該催化劑需要使電子、空氣、電解質水溶液的供給路徑分別連接在一起,以便產生涉及電極反應的空氣(氧)、水、電子、oh-的授受。水和oh-的供給路徑由於為電解質水溶液,因而電解質水溶液需要從隔膜側滲入催化劑層4,正極3的至少隔膜對置側必須是親水性且多孔質的。另一方面,正極的相反側的面雖然為向空氣開放的結構,但為了設計為避免強鹼性的電解質水溶液從空氣開放口漏出且空氣能夠擴散的結構,空氣極的空氣開放側要求為疏水性且多孔質的。
為了滿足以上的結構上的要件,通常設計為如下的兩層結構:從隔膜對置側(負極對置側)開始,設置以碳材料和催化劑成分為主成分的親水性且多孔質的催化劑層4,繼而設置疏水性的多孔質層5;用於與外部電路授受電子的正極側的集電體8通常使用金屬網,與疏水性多孔質層的空氣開放側接觸配置(圖2),或者在催化劑層和疏水性多孔質層之間配置(圖1)。
這種結構的正極為了進行效率良好的電極反應,重要的是確保電極反應所需要的物質向配置於電極中的催化劑的移動路徑,從而促進物質移動。從更微觀的視點上看,在可以與外部電路導通並供給反應所需要的電子的碳材料網絡、和由空氣可以從電池外部的大氣擴散開來的孔隙連接而成的空氣擴散網絡、和可以根據充放電而與負極授受oh-的電解質水溶液網絡的接點即三相界面,需要儘可能多地存在作為反應場的催化劑。
為了形成這樣的三相界面,一直以來提出了幾種方法。在由親水性催化劑層和疏水性多孔質層組合而成的通常的兩層結構中,如果在用親水性材料形成的多孔質催化劑層中滲入電解質水溶液,則電解質水溶液滲入形成於催化劑層的整個氣孔中,實質上存在電解質水溶液和空氣的三相界面局限於親水性催化劑層和疏水性多孔質層的界面部分9(圖3)。也就是說,配置於催化劑層內部的催化劑成分的大部分被電解質水溶液包圍,不能供給電極反應所需要的空氣(氧)而變得不會有助於反應。
為了避免這樣的狀況,作為正在廣泛而通常地進行的手段,可以列舉出在用親水性碳材料形成的催化劑層中複合作為憎水成分的ptfe的方法。另外,作為類似的方法,專利文獻1提出了在催化劑層中複合作為疏水成分的蠟的方法。在催化劑層中複合作為疏水性的ptfe或蠟的方法的目標是:通過在親水性多孔質催化劑層的一部分上形成疏水部分,便在催化劑層內形成空氣(氧)的擴散路徑,從而使三相界面增加。
另外,作為其它方法,專利文獻2提出了一種對親水性多孔質催化劑層和疏水性多孔質層的界面實施紋理加工、從而賦予界面以凹凸形狀的電極。本方法的目標是:通過增加被電解質水溶液充滿的親水性多孔質催化劑層和疏水性多孔質層的界面的面積,使形成於界面部分的三相界面增加。
再者,作為其它方法,專利文獻3提出了一種將多孔質催化劑層用擔載著催化劑成分的親水性多孔質材料和疏水性多孔質材料的混合物形成的方法。作為基本的構思,本方法也與複合ptfe或蠟的方法同樣,通過在親水性多孔質催化劑層中複合疏水性材料,便形成空氣的擴散路徑,使三相界面增大,從而謀求電極反應的效率化。
另一方面,即便是與金屬空氣電池同樣,活性物質為氧,配置氧還原催化劑的燃料電池的空氣極的催化劑層,也進行親水性-疏水性的材料控制。在燃料電池的空氣極(正極)的催化劑層中,重要的是使存在於由活性物質的空氣中的氧能夠擴散的孔隙、作為質子的傳遞路徑的電解質材料以及能夠傳遞電子的碳材料形成的三相界面的催化劑高密度化。一般地說,作為質子傳遞路徑的電解質材料在高溼潤條件下質子傳遞阻力達到最小,因而需要將催化劑層構成材料設計成親水性,使電解質材料保持在溼潤狀態,從而抑制質子傳導阻力的上升。
另一方面,通過空氣極反應而產生水,為了抑制因該水引起的氣體擴散路徑的封閉,需要在催化劑層中複合疏水性材料而確保含有作為活性物質的氧的空氣的擴散路徑。滿足這兩個相反要求的高度的親水性-疏水性的材料控制特別要求以高電流密度進行發電的高性能催化劑層。從這樣的角度考慮,專利文獻4提出了在催化劑層中包含具有疏水性且對氣體擴散有利的立體結構的碳黑的方法。再者,作為兼顧使電解質材料保持在溼潤狀態從而不會損害質子傳導路徑即電解質材料的質子傳導性、且確保氣體擴散路徑的技術,專利文獻5提出了使疏水性碳材料成為凝聚結構而分散於催化劑層中的技術。
專利文獻6公開了一種固體高分子型燃料電池用電極,其通過將相對溼度在90%下的水蒸氣吸附量為1ml/g~20ml/g的碳材料用作氣體擴散碳材料,可以有效地抑制因水引起的氣體擴散路徑的封閉,並能夠以穩定的電壓取出電流。
另外,在專利文獻7中,公開了在將燃料電池用催化劑粉末的粒度分布的體積累積頻率分別達到50%以及90%時的粒徑各自設定為d50以及d90時,通過滿足(d90/d50)≤2.5的條件,使電極中的氣體擴散性得以提高。
另外,在專利文獻8中,為了提供對用作氣體擴散電極合適的材料,公開了一種由包含多中孔性納米結構疏水性材料的第1層、和包含在第1層上配置的多中孔性納米結構親水性材料的第2層構成的電極。
現有技術文獻
專利文獻
專利文獻1:日本特表2008-502118
專利文獻2:日本特表2003-514367
專利文獻3:日本特表2012-502427
專利文獻4:日本特開2007-273145
專利文獻5:日本特開2009-252359
專利文獻6:國際公開wo2010/047415
專利文獻7:日本特開2008-147007
專利文獻8:日本特表2012-502427
技術實現要素:
發明所要解決的課題
ptfe的複合或專利文獻1提出的蠟的複合雖然通過設置使電解質水溶液不會滲入催化劑層中的疏水部分而確保空氣的擴散路徑,可以期待一定的效果,但複合的材料實質上為絕緣體,因而成為課題的是難以調整電子傳導性和疏水性的平衡。具體地說,如圖5所示,為了充分確保空氣的擴散路徑,如果過於增加疏水性的ptfe或者蠟10在催化劑層中的含量,則電子傳導阻力增大。另一方面,如圖4所示,為了抑制電子傳導阻力的上升,如果過於降低ptfe或者蠟10的含量,則空氣的擴散性變得不充分。特別地,為了取出大電流密度,單憑ptfe或蠟的量的加減,難以發揮可以令人滿意的性能。
專利文獻2提出的催化劑層和疏水性多孔質層的界面紋理加工雖然三相界面的面積在幾何學上有一定量的增大(圖6),但能夠期待三相界面的部位局限於催化劑層和疏水性多孔質層的界面,畢竟不能成為有效利用催化劑層內部的催化劑成分的技術。
專利文獻3提出的混合疏水性多孔質材料的方法可以看到對三相界面的面積增大具有相當的效果。但是,在單純混合的方法中,將產生與ptfe的複合和專利文獻1的情況同樣的問題。也就是說,如圖7所示,如果疏水性多孔質材料的含量較少,則催化劑層內部的疏水性多孔質材料11被親水性多孔質材料包圍。被親水性多孔質材料包圍的疏水性多孔質材料11實質上不會作為空氣的擴散路徑發揮作用,從而在獲得高電池性能方面是不充分的。另一方面,如圖8所示,如果增加疏水性多孔質材料11的含有率而使疏水性多孔質材料11成為連結結構,則可能產生被疏水性多孔質材料11包圍的親水性多孔質材料。其結果是,由於產生電解液不能到達催化劑成分的部位,因而不能獲得高電池性能。
特別地,為了獲得高輸出密度,需要一定量以上的催化劑量,但除此以外,提高含量而使疏水性多孔質材料成為連結結構,由此催化劑層的厚度增大到必要以上,其結果是,電子傳導路徑、空氣擴散路徑等各路徑長度增大,結果招致與物質移動阻力相伴的ir損耗的增大,從而成為效率低下的原因。另外,在專利文獻3中,雖然根據極性基的有無而區分親水性多孔質材料以及疏水性多孔質材料,但難以確保完全沒有極性基的材料,除此以外,即便是具有極性基的材料,也根據其含量的不同而存在實質上顯示出疏水性的材料,在該文獻所記載的方法中,設計可以穩定地表現出高性能的催化劑層是困難的。
另外,在燃料電池領域提出的專利文獻4~6的方法中,除了具有疏水性碳材料自身發達的立體結構且容易成為空氣的擴散路徑以外,疏水性碳材料還形成凝聚體,因而即使在金屬空氣電池的領域,也可以期待能夠以較少的疏水性碳材料複合量確保氣體擴散路徑的優良的效果(圖9、圖11)。此外,圖11是放大表示專利文獻5所公開的催化劑層的概略構成的說明圖。專利文獻5所公開的催化劑層可區分為催化劑凝聚相16和氣體擴散凝聚相17。催化劑凝聚相16由擔載催化劑成分的碳材料12、電解質材料13以及導電助劑碳材料14構成。氣體擴散凝聚相17由氣體擴散碳材料15構成。圖9的疏水性多孔質材料11與圖11的氣體擴散碳材料15相當。如圖11所示,在專利文獻5中,催化劑凝聚相16成為連續體,氣體擴散凝聚相17分散於催化劑凝聚相16內。但是,在電解質水溶液沿著親水性碳材料即催化劑載體而滲入催化劑層的金屬空氣電池中,如圖9和圖12所示,三相界面的形成可以期待的部位9局限於親水相和疏水相的界面。因此,越是適用於燃料電池的情況,越不能有效利用親水相中的催化劑,為了獲得高電池性能,在金屬空氣電池的用途時,其課題是需要特殊的設計。
再者,在專利文獻4和5中,作為用於限定擔載催化劑成分的碳材料的指標,規定了相對溼度在90%下的水蒸氣吸附量。相對溼度在90%下的水蒸氣吸附量可以說是一個定量的指標。但是,該指標是用於確保可以在親水性的碳材料表面保持的水份量(換算成水蒸氣量)、即在親水性碳材料的附近配置的電解質材料的溼潤環境的指標。該指標是燃料電池領域特有的指標。另一方面,在滲入電解質水溶液而工作的金屬空氣電池中,重要的是碳材料表面對電解質水溶液的潤溼性。因此,可以保持碳材料的水份量即本指標難以說是適當的。
另一方面,在專利文獻4~6中,作為用於限定沒有擔載催化劑成分的氣體擴散碳材料的指標,規定了相對溼度在90%下的水蒸氣吸附量。相對溼度在90%下的水蒸氣吸附量可以說是一個定量的指標。但是,該指標是用於確保在燃料電池工作的環境下具有作為燃料的氣體可以擴散這種程度的疏水性的碳材料的指標。該指標是燃料電池領域特有的指標。另一方面,在滲入電解質水溶液而工作的金屬空氣電池中,重要的是碳材料表面對電解質水溶液的潤溼性。因此,本指標難以說是適當的。
另外,在專利文獻7~8中,與電極的催化劑層材料所要求的親水性和疏水性有關的特性沒有以定量的方式進行定義。因此,在專利文獻7~8所公開的技術中,意圖增大金屬空氣電池在正極中的三相界面的面積是困難的。
這樣一來,在專利文獻1~8所公開的技術中,難以擴大金屬空氣電池在正極中的三相界面(換句話說,在三相界面配置較多的催化劑)。另外,即使擴大三相界面,也由於損害電子傳導性等原因,不能獲得高電池特性。
於是,本發明是鑑於上述問題而完成的,本發明的目的在於提供一種可以在三相界面配置較多的催化劑、進而能夠改善電池特性、新穎且得以改良的金屬空氣電池。
用於解決課題的手段
發明人為解決上述課題,鑑於電解質水溶液滲入催化劑層而發揮作用的金屬空氣電池的作用機理,根據如下等指針而對金屬空氣電池的作為正極催化劑層最合適的結構進行了研究,所述指針包括:1)儘可能增加存在於三相界面的催化劑;2)將疏水性材料設定為碳材料,從而不會因導入的疏水性材料而損害催化劑層中的電子傳導性;3)設計為可以將催化劑層中的疏水性材料的導入量抑制在最低限度的結構,從而不會增大電子傳導路徑、空氣擴散路徑、oh-離子的傳播路徑等各路徑的長度。
其結果是,如圖10和圖13所示,本發明人構想將催化劑層的結構分為以擔載著催化劑成分的親水性碳材料(碳材料a)18為主體的親水凝聚相(凝聚相x)20、和以不含有催化劑成分的疏水性碳材料(碳材料b)19為主體的疏水凝聚相(凝聚相y)21這兩相。而且將親水凝聚相20設定為連續結構,並將疏水凝聚相21設計為分散於親水凝聚相的連續結構中的結構。再者,在親水凝聚相20內部微細分散著疏水性碳材料19。其結果是,成功地以從前沒有的程度形成了三相界面。
接著,本發明人著眼於微孔(直徑為2nm以下的孔)的表面積在擔載催化劑成分的親水性碳材料的總表面積中所佔的比例。具體地說,本發明人著眼於微孔的表面積在親水性碳材料的總表面積中所佔的比例不處於支配地位的情況和處於支配地位的情況。
在微孔的表面積於親水性碳材料的總表面積中所佔的比例處於支配地位的情況下,如果微孔過深,則空氣和電解液難以到達配置於微孔深部的催化劑。因此,如果微孔過深,則成為效率低下的金屬空氣電池。於是,本發明人在微孔表面積的比例較高的親水性碳材料的情況下,通過減小平均粒徑(平均粒子直徑),使微孔不會極端加深。
另一方面,在微孔的表面積於親水性碳材料的總表面積中所佔的比例不處於支配地位的情況下,可以使電解質水溶液與擔載著親水性碳材料的催化劑成分接觸,而且可以使空氣高效地擴散。由此,可以使存在於三相界面的催化劑以更高的密度存在。
再者,本發明人也著眼於親水性碳材料的粒徑分布(粒子直徑分布)。也就是說,如果粒徑分布過寬,則粒徑較小的親水性碳材料有可能堵塞催化劑層。在此情況下,空氣難以擴散至催化劑層內。於是,本發明人使催化劑層碳材料的粒徑分布儘可能變得尖銳。由此,可以使存在於三相界面的催化劑以更高的密度存在。
接著,本發明人考慮到與金屬空氣電池的空氣極的催化劑層材料所要求的親水性和疏水性有關的特性為對電解質水溶液的潤溼性,對於催化劑層材料所要求的親水性和疏水性,研究了可以定量判斷的適當的定量指標。其結果是,本發明人發現這樣的指標是相對壓力(relativepressure)為0.1時的水蒸氣吸附量。通過以相對壓力是0.1時的水蒸氣吸附量為指標,使穩定地獲得高性能的催化劑層的設計成為可能。
此外,在專利文獻5的實施例9中,公開了與圖10和圖13所示的催化劑層類似的催化劑層。也就是說,即使在專利文獻5的實施例9所公開的催化劑層中,親水凝聚相也包含擔載著催化劑成分的親水性碳材料、和水蒸氣吸附量比較低的碳材料即疏水性碳材料。而且對於擔載著催化劑成分的親水性碳材料,其立體結構並不發達,為相對於總表面積的直徑為2nm以下的微孔表面積處於絕對的支配地位且極其容易保持水的特殊的碳材料。在擔載著催化劑的親水性碳材料使用微孔不處於支配地位的碳材料的情況下,將招致在催化劑層內共存的電解質材料的乾燥。也就是說,在實用的燃料電池的發電條件即乾燥條件下,將招致發電性能顯著劣化的結果。因此,在專利文獻5的實施例9中,作為親水性碳材料,使用微孔處於絕對支配地位的碳材料。
但是,在專利文獻5的實施例9中,對於親水性碳材料的平均粒徑和粒徑分布沒有做任何考慮。因此,在專利文獻5的實施例9中,有微孔過深的可能性。另外,有可能因粒徑較小的親水性碳材料粒子而使催化劑層發生堵塞。再者,專利文獻5以與相對壓力為0.1時的水蒸氣吸附量完全不同的指標即相對溼度在90%下的水蒸氣吸附量為基礎而對碳材料進行了評價。這樣一來,本發明與專利文獻5的實施例9所公開的構成完全不同。
也就是說,本發明包含以下的構成。
(1)一種金屬空氣電池用電極,其特徵在於:金屬空氣電池的空氣極的催化劑層包含催化劑成分以及碳材料;
所述碳材料由擔載著所述催化劑成分的碳材料a、和沒有擔載所述催化劑成分的碳材料b這2種構成;
所述催化劑層由合計含有超過50質量%的所述催化劑成分、所述碳材料a以及所述碳材料b的凝聚相x、和含有50質量%的所述碳材料b的凝聚相y構成;
所述凝聚相x為連續體,所述凝聚相y為分散於所述凝聚相x中的結構;
所述碳材料a滿足以下的特徵(i)或者(ii);
所述碳材料b在25℃且相對壓力為0.1的環境下,水蒸氣吸附量低於0.1cm3/g,dbp吸油量(x)(cm3/100g)與基於bet評價的比表面積(sbet)之比(x/sbet)為0.5以上;
(i)所述碳材料a在25℃且相對壓力為0.1的環境下,水蒸氣吸附量為0.1cm3/g以上,採用氮吸附等溫線的t圖分析求出的微孔表面積smicro與總表面積stotal之比smicro/stotal為0.90以上,平均粒徑d50低於1.5μm,d90和d10的差(d90-d10)與d50之比(d90-d10)/d50低於1.0;
(ii)所述碳材料a在25℃且相對壓力為0.1的環境下,水蒸氣吸附量為0.1cm3/g以上,採用氮吸附等溫線的t圖分析求出的微孔表面積smicro與總表面積stotal之比smicro/stotal為0.90以下。
(2)根據上述(1)所述的金屬空氣電池用電極,其特徵在於:所述碳材料a在25℃且相對壓力為0.1的環境下,水蒸氣吸附量為0.1cm3/g以上,採用氮吸附等溫線的t圖分析求出的微孔表面積smicro與總表面積stotal之比smicro/stotal為0.90以上,平均粒徑d50低於1.5μm,d90和d10的差(d90-d10)與d50之比(d90-d10)/d50低於1.0;
所述碳材料b在催化劑層中的含量超過10質量%且低於50質量%。
(3)根據上述(1)所述的金屬空氣電池用電極,其特徵在於:所述碳材料a在25℃且相對壓力為0.1的環境下,水蒸氣吸附量為0.1cm3/g以上,採用氮吸附等溫線的t圖分析求出的微孔表面積smicro與總表面積stotal之比smicro/stotal為0.90以下;
所述碳材料b在催化劑層中的含量為5質量%以上且低於50質量%。
(4)根據上述(1)~(3)中任一項所述的金屬空氣電池用電極,其特徵在於:在所述催化劑層的斷面的10μm×10μm的視場中,當量圓直徑為300nm以上這種大小的沒有擔載催化劑成分的碳材料凝聚相存在1個以上。
(5)根據上述(1)~(4)中任一項所述的金屬空氣電池用電極,其特徵在於:所述催化劑層中的在金屬極對置側的所述碳材料b的含有率α為0質量%以上且低於20質量%;
所述催化劑層中的在空氣開放側的所述碳材料b的含有率β超過10質量%且低於50質量%;
且滿足α<β。
發明的效果
正如以上所說明的那樣,根據本發明,可以在三相界面配置較多的催化劑,進而能夠改善電池特性。
附圖說明
圖1是表示以前的金屬空氣電池的概略構成的說明圖。
圖2是表示以前的金屬空氣電池的概略構成的說明圖。
圖3是表示以前的金屬空氣電池正極的概略構成的說明圖。
圖4是表示以前的金屬空氣電池正極的概略構成的說明圖。
圖5是表示以前的金屬空氣電池正極的概略構成的說明圖。
圖6是表示以前的金屬空氣電池正極的概略構成的說明圖。
圖7是表示以前的金屬空氣電池正極的概略構成的說明圖。
圖8是表示以前的金屬空氣電池正極的概略構成的說明圖。
圖9是表示將以前的燃料電池的催化劑層用於金屬空氣電池時(電解質水溶液滲入時)的正極的概略構成的說明圖。
圖10是表示本發明的催化劑層的概略構成的說明圖。
圖11是表示以前的燃料電池的催化劑層的概略構成的說明圖(放大圖)。
圖12是表示將以前的燃料電池的催化劑層用於金屬空氣電池時(電解質水溶液滲入時)的催化劑層的概略構成的說明圖(放大圖)。
圖13是表示本發明的催化劑層的概略構成的說明圖(放大圖)。
圖14是表示本發明的催化劑層的概略構成的說明圖(放大圖)。
圖15是表示本發明的擔載著催化劑成分的親水性碳材料(碳材料a)的概略構成的說明圖(放大圖)。
具體實施方式
下面參照附圖,就本發明優選的實施方式進行詳細的說明。此外,在本說明書以及附圖中,對於具有實質上相同的功能構成的構成要素,通過標註相同的符號而省略其重複說明。
[本發明的金屬空氣電池的空氣極的催化劑層的結構]
關於構成本發明的催化劑層結構,圖10以及圖13示出了示意圖。在所有的圖中,由於是示意性地表現各材料和凝聚相,因而各材料的形狀和相對尺寸與實際情況不同。本發明的金屬空氣電池用電極中含有的催化劑層由包含催化劑成分和碳材料的混合物構成。所述碳材料由擔載著所述催化劑成分的碳材料a(符號18)、和沒有擔載所述催化劑成分的碳材料b(符號19)這2種構成。所述催化劑層由以所述碳材料a和碳材料b為主成分的凝聚相x(符號20)、以及以碳材料b為主成分的凝聚相y(符號21)這2種凝聚相構成,具有在作為連續體的凝聚相x中分散有凝聚相y的結構。
如果電解質水溶液6滲入圖13所示的催化劑層中,則如圖14所示,可以形成能夠期待三相界面的形成的、電解質水溶液與空氣的界面9。在本實施方式中,由於在凝聚相x內分散著碳材料b(疏水性碳材料),因而電解質水溶液與空氣的界面9的面積增大。其結果是,可以在三相界面配置較多的催化劑成分。此外,擔載著催化劑成分的碳材料a(圖13以及圖14中的符號18)如圖15所示,成為催化劑成分23固定(以下也稱為「擔載」)於碳材料a(圖15的符號22)的表面以及微孔內的複合材料。
此外,詳細情況後述,而在本發明的一實施方式中,碳材料a的微孔的表面積在總表面積中所佔的比例處於支配地位。在這樣的實施方式中,較多的催化劑成分23擔載於碳材料a的微孔內。
在此,凝聚相x以催化劑成分、碳材料a以及碳材料b為主成分。具體地說,催化劑成分、碳材料a以及碳材料b的總質量相對於凝聚相x的總質量超過50質量%。另外,凝聚相y以碳材料b為主成分。具體地說,凝聚相y相對於凝聚相y的總質量含有超過50質量%的碳材料b。
本發明中的所謂凝聚,是指催化劑成分的一次粒子和碳材料的單一粒子因範德瓦爾斯力和庫侖力等粒子間力而集聚的狀態,將催化劑層中形成的這種狀態的催化劑成分的一次粒子和碳材料的單一粒子的一個塊的塊狀體(凝聚體)稱為凝聚相。本發明的各成分各自具有作為催化劑層的成分所需要的功能。如果為催化劑成分,則是作為催化劑的功能,如果為碳材料,則是作為電子傳導體的功能。特別地,碳材料在本發明中分別使用碳材料a和碳材料b這2種碳材料,它除了作為電子傳導體的功能以外,如果為碳材料a,則具有擔載催化劑成分的功能和使電解質水溶液滲入催化劑層中的功能,如果為碳材料b,則具有使空氣高效地擴散的功能。通過設計為本發明的催化劑層所具有的兩種凝聚相結構,可以使催化劑成分、碳材料a以及碳材料b所具有的功能有效地表現出來,從而能夠以高水平形成金屬空氣電池的正極催化劑層所需要的三相界面。
第一,碳材料b通過以凝聚相y的形式作為一個塊的塊狀體而分散於催化劑層中,便可以最大限度地活用碳材料b所具有的表面特性和立體結構,從而在催化劑層中容易連續地形成空氣的移動路徑。也就是說,在碳材料b中,單一粒子本身具有發達的立體結構,在該立體結構中具有空間,通過使碳材料b彼此之間凝聚,便有可能使立體結構中的空間以三維的方式連結起來,從而能夠以比將單一粒子分散於催化劑層中的情況更少的量,使空氣容易向催化劑層內擴散的粗的路徑變得發達。
第二,在本發明的催化劑層中,通過在與電解質水溶液具有親和性的碳材料a上擔載催化劑成分並包含在一個凝聚相x中,從而在催化劑層中形成連續體,便可以使電解質水溶液與催化劑層中含有的大概所有的催化劑成分接觸。也就是說,與電解質水溶液具有親和性的碳材料a在催化劑層內形成連續體,因而從催化劑層的隔膜側接觸的電解質水溶液沿著碳材料a的表面而滲入催化劑層,從而可以滲入到催化劑層的整個區域。
在本發明的一實施方式中,碳材料a的微孔的表面積在總表面積中所佔的比例處於支配地位。在這樣的實施方式中,催化劑成分主要擔載於傳遞電解質溶液的碳材料a的微孔內,但由於微孔本身為較淺的結構,因而電解質水溶液也可以容易地與微孔內的催化劑成分接觸。
另外,在本發明的其它實施方式中,作為碳材料a,也可以選擇微孔的表面積未處於支配地位的碳材料。在該其它實施方式中,催化劑成分擔載於傳遞電解質溶液的碳材料a的表面,因而電解質水溶液和催化劑成分能夠有效地接觸。
第三,通過使碳材料b微細分散於存在擔載著催化劑成分的碳材料a的凝聚相x中,便可以向催化劑層中含有的催化劑成分有效地供給空氣中的氧。在凝聚相x中未含有與電解質水溶液不具有親和力的碳材料b的情況下,滲入催化劑層中的電解質水溶液與從大氣擴散來的空氣的界面局限於凝聚相x和凝聚相y的界面部分,其結果是,進行放電反應的催化劑成分也局限於界面部分。但是,通過使碳材料b微細分散於凝聚相x中,便能夠在凝聚相x中形成雖然微細但空氣可以擴散的孔,從而能夠使沿著由凝聚相y形成的粗的路徑擴散而來的空氣進一步高效地擴散至存在於凝聚相x中的內部的催化劑成分。
在本發明的催化劑層中,催化劑層的在負極對置側的碳材料b的含有率α優選為0質量%以上且低於20質量%,催化劑層的在空氣開放側中含有的碳材料b的含有率β優選為超過10質量%且低於50質量%。如果滿足α<β,則在催化劑層中,滲入催化劑層的電解質水溶液與擴散的空氣的界面面積有增大的傾向,因而是優選的。
催化劑層既可以是兩層以上的多層結構,也可以是負極對置側的第一層的碳材料b的含有率α為0質量%以上且低於20質量%、且空氣開放側的第二層的碳材料b的含有率β超過10質量%且低於50質量%的結構。另外,催化劑層也可以是如下的結構:在第一層和第二層之間夾持著單層或者多層的碳材料b的含有率設定在第一層和第二層中間的層。
或者,催化劑層也可以是如下的結構:沒有明確的層結構,催化劑層中含有的碳材料b的含有率從負極對置側向大氣開放側連續地發生變化。但是,在為碳材料b的含有率連續地發生變化的結構的情況下,如果碳材料b的含有率α為20質量%以上,則滲入催化劑層中的電解質水溶液的網絡往往變得貧乏,從而有時難以獲得電解質水溶液和空氣的界面增大效果。另外,如果含有率β為10質量%以下,則催化劑層中的碳材料b的網絡具有局限於催化劑層的大氣開放側的傾向,如果為50質量%以上,則在催化劑層的大氣開放側,碳材料b往往不會成為分散結構,因而是不優選的。另外,如果α在β以上,則有時產生電解質水溶液和空氣的網絡在催化劑層中變得貧乏的部位,因而是不優選的。
本發明的催化劑層的主成分為催化劑成分、碳材料a以及碳材料b,但為了補足凝聚相x和凝聚相y各自的凝聚結構,且提高催化劑層的機械強度,可以含有粘結劑材料。粘結劑材料無論是用於凝聚相x和凝聚相y任一方還是用於兩方都沒有關係。優選的粘結劑材料為高分子材料。只要是將凝聚相x或者凝聚相y各自中含有的成分連結固定而補足凝聚結構的材料,就沒有特別的限定。如果舉出特別優選的例子,聚四氟乙烯或者在聚四氟乙烯的主骨架上連接著具有官能團的支鏈這些結構的全氟碳類的高分子由於難以發生通過化學反應進行的改性,且將對電極反應的影響抑制在最小限度的水平,因而是優選的。
催化劑層中的粘結劑材料的含有率優選為0質量%以上且低於30%。只要在該範圍,就可以補足本發明所意圖的凝聚結構的形成,從而可以提高催化劑層的機械強度。這樣的高分子大多為絕緣體,即使具有導電性,也大多為比碳材料的電導性難以進行電導的材料,因而如果為30質量%以上,則進入碳材料彼此之間的接觸界面而分斷電子傳導路徑,從而往往使整個催化劑層的導電性下降,成為ir損耗而使電池性能降低。
[凝聚相x的結構]
本發明的催化劑層中含有的凝聚相x的結構優選為如下的結構:擔載著催化劑成分的碳材料a成為連續結構,在該連續結構中微細分散有碳材料b。在催化劑層中形成有連續體的凝聚相x的更縱深通過採取這樣的結構,碳材料a可以遍及催化劑層的整個區域而取得連續的結構。其結果是,從催化劑層外部開始接觸的電解質水溶液在碳材料a的表面傳播,從而能夠潤溼催化劑層中的所有碳材料a的表面,以致能夠與擔載於碳材料a上的催化劑成分高效地接觸。另一方面,在凝聚相x所含有的碳材料b中,碳材料b所具有的立體結構中的空間在凝聚相x中形成微細的空氣擴散路徑。電極外部的空氣在催化劑層中,可以通過由凝聚相y形成的粗的空氣擴散路徑而向催化劑層內部擴散,進而可以在凝聚相x中通過由碳材料b形成的微細的擴散路徑而供給至凝聚相x中含有的催化劑成分。由此,可以使空氣有效地向凝聚相x中含有的催化劑成分擴散,其結果是,可以有效利用催化劑層中含有的催化劑成分。
因此,本發明的凝聚相x中含有的碳材料b優選的含有率只要是至少在凝聚相x中不會分斷碳材料a的連續結構這樣的程度,就沒有限定。更優選的碳材料b在凝聚相x中的含有率以抽取出催化劑成分的質量比(碳材料b的質量)/(碳材料a的質量+碳材料b的質量)計,為0.1~0.5。如果低於0.1,則具有難以看到在凝聚相x中微細分散著碳材料b的效果的傾向。如果超過0.5,則在凝聚相x內部,碳材料b有時包圍著碳材料a,電解質水溶液有可能不能與擔載於碳材料a上的催化劑成分接觸而不能獲得充分的電池性能。
[凝聚相y的結構]
本發明的催化劑層的結構可以通過觀察其斷面而進行確認。其方法是在催化劑層的任意場所以任意角度製作切斷面,對其斷面進行觀察,從而確認沒有擔載催化劑成分的碳材料形成了凝聚體(凝聚相)。所述凝聚體與本發明的凝聚相y相對應。
在催化劑層斷面的10μm×10μm面積的視場中,優選至少分散1個當量圓直徑為300nm以上大小的沒有催化劑成分的碳材料凝聚相(凝聚相y)。在低於1個時,由於催化劑層形成時平均混合碳材料a和碳材料b、或者沒有擔載催化劑成分的碳材料b的含有率過低,因而至少碳材料b沒有形成本發明意圖的凝聚相y,所以在該催化劑層中,空氣的傳遞路徑不發達,空氣的擴散性差,從而不能表現出特別穩定的放電性能。在該視場中,更優選至少存在1個當量圓直徑為500nm以上大小的沒有催化劑成分的碳材料凝聚相(凝聚相y)。如果為上述結構,則至少可以抑制放電性能變得不穩定,從而獲得穩定的放電性能。
催化劑層的斷面的形成方法並沒有特別的限定,例如可以列舉出用刀片或剪刀將催化劑層切斷的方法等。在催化劑層不含有粘結劑成分、或者粘結劑成分量較少而使催化劑層較脆的情況下,也可以使用環氧等樹脂進行補強而將其切斷,以便使催化劑層的結構不會遭到破壞。特別優選的方法是使用冷凍切片機(cryo-microtome)等,在用液氮冷凍的環境下形成催化劑層的切斷面的方法。在該方法中,將作為試料的催化劑層設置在冷凍切片機上,使用由金剛石或玻璃製成的修整刀對催化劑層表面進行切削,並對生成的切削麵進行觀察。
在對催化劑層的斷面進行觀察的方法中,可以用二次電子像和背散射電子圖像這兩者對同一視場進行觀察,優選為能夠以至少1萬倍以上的放大倍數進行觀察的掃描型電子顯微鏡。二次電子像可以反映催化劑層斷面的凹凸信息,從而可以確認碳材料或氣孔的存在。如果採用高精度的電子顯微鏡,則可以確認催化劑成分的存在,如果觀察同視場的背散射電子圖像,則可以反映成分的分布信息,例如在催化劑成分使用金屬的情況下,則產生催化劑成分為明、沒有催化劑成分的地方為暗的對比度而得到圖像。如果對本發明的催化劑層的二次電子像和背散射電子圖像進行比較,則在同視場中,儘管在二次電子像中存在碳材料,但在背散射電子圖像中可以看到暗的對比度的部分、即不存在催化劑成分的碳材料。如果所述部分、即不具有催化劑成分的碳材料部分的外周的當量圓直徑在300nm以上,則成為本發明優選的方式。
下面對能夠更定量地識別當量圓直徑在300nm以上這種大小的沒有催化劑成分的碳材料凝聚相(凝聚相y)的存在的例子進行敘述。對於背散射電子圖像,在1萬倍的放大倍數下採用272dpi×272dpi以上的解析度、且以256個等級的層次(hierarchy)採集亮度。對於採集的圖像的亮度,使用圖像分析軟體進行二值化,從而將從暗的至第110層次的範圍用黑色表示,並使從第111層次向明的直至第256層次的範圍成為白色。在這樣的狀態下,黑色點孤立為島狀的點大量產生,因而為了使目標的範圍明確化,對各黑色點進行1次膨脹處理,從而對相鄰的點彼此之間進行識別。再者,執行填孔處理,對範圍內的空白部分進行填孔,從而識別為同一範圍。最後,進行使膨脹的部分復原的收縮處理(degenerationprocess),從而使目標的範圍明確化。而且由各黑色部分的面積算出各黑色部分的當量圓直徑,並將低於300nm的部分全部切除。在剩餘的黑色部分中,在二次電子像中存在碳材料的情況成為本發明優選的方式。
在本發明中,不必採用上述所有的分析手法對沒有催化劑成分的碳材料凝聚相(凝聚相y)進行觀察來滿足本發明的規定範圍,只要採用1種分析手法得到的值滿足本發明的規定範圍,就可以得到其效果。
[本發明所使用的催化劑成分]
本發明所使用的催化劑成分只要是在催化劑成分上至少進行氧還原反應的成分,就沒有限定。只要至少進行氧還原反應,就可以表現出作為一次電池的功能。在作為二次電池發揮作用的情況下,可以使用一併具有在催化劑成分上進行氧生成反應的功能的成分,或者複合使用進行氧還原反應的成分和進行氧生成反應的成分。作為優選的催化劑成分的例子,可以列舉出鉑、鈀、釕、金、銀、銠、鋨、銥、鎳、鐵、鈷、鉬、錳等金屬,使2種以上的這些金屬複合化的金屬的複合體或合金,這些金屬與有機化合物或無機化合物的絡合物、以及金屬氧化物。另外,也可以使用將上述的2種以上複合而成的材料等。
[本發明所使用的碳材料]
所述碳材料a、b都需要具有電子傳導性,但不優選由與空氣、電解質水溶液和其它電池構成材料發生化學反應,或者通過與電解質水溶液的接觸而使碳材料中含有的物質溶出的碳材料形成。在本發明中,優選使用化學性質穩定的碳材料。作為這樣的碳材料,可以例示出碳黑、石墨、碳纖維、活性炭等或它們的粉碎物、碳納米纖維、碳納米管、石墨烯等碳化合物等。可以將前述優選的碳材料的1種或者2種以上混合而用於所述碳材料a、b的原材料。另外,所述碳材料也可以是使用各種鑄型而形成了結構的形狀。
[碳材料a]
在本發明所使用的碳材料中,碳材料a除了擔載著催化劑成分的功能以外,還具有使電解質水溶液滲入催化劑層中的功能。為了使電解質水溶液容易沿著碳材料a的表面而滲入催化劑層中,優選提高碳材料a和電解質水溶液的潤溼性。該潤溼性可以用一定條件下的水蒸氣吸附量來判斷。為了更有效地表現出該功能,如果碳材料a選擇在25℃、相對溼度10%下(即相對壓力0.1下)的水蒸氣吸附量為0.1cm3/g以上的碳材料,則與電解質水溶液的親和性升高,從而容易使電解質水溶液沿著碳材料a的表面而滲入催化劑層中。其結果是,可以使擔載於碳材料a的表面的催化劑成分與電解質水溶液高效地接觸,從而可以有效地授受為使催化劑成分發揮作用所需要的oh-離子。因此,本發明的碳材料a對於電解質水溶液越容易潤溼越好,在25℃、相對溼度10%下的水蒸氣吸附量的上限值並沒有特別的限制。如果例示出25℃、相對溼度10%下的水蒸氣吸附量的實質性的上限值,則可以列舉出一般認為用非常高比表面積的活性炭可以得到的10cm3/g左右。
此外,碳材料a在25℃、相對溼度10%下的水蒸氣吸附量如果低於0.1cm3/g,則電解質水溶液往往不會滲入催化劑層中,在催化劑層中不能與電解質水溶液接觸的催化劑成分變得容易產生,因而往往不會進行充分的電極反應。25℃、相對溼度10%下的水蒸氣吸附量如果選擇0.2cm3/g以上的碳材料,則不管碳材料的表面積和立體結構如何,可以使電解質水溶液更加切實地滲入催化劑層中,從而大部分催化劑成分可以與電解質水溶液接觸,因而能夠有效地發揮催化劑成分的功能。
25℃、相對溼度10%下的水蒸氣吸附量是將置於25℃的環境下的每1g碳材料所吸附的水蒸氣量換算成標準狀態的水蒸氣體積來表示。25℃、相對溼度10%下的水蒸氣吸附量的測定可以使用市售的水蒸氣吸附量測定裝置來測定。
作為所述碳材料a,可以使用微孔的表面積在總表面積中所佔的比例處於支配地位的碳材料、或者在總表面積中所佔的微孔的表面積未處於支配地位的碳材料之中的至少任1種。所述碳材料a也可以混合使用在總表面積中所佔的微孔的表面積處於支配地位的碳材料、和在總表面積中所佔的微孔的表面積未處於支配地位的碳材料。
但是,對於擔載在碳材料a上的催化劑成分,為了使電解質水溶液與之接觸,而且使空氣高效地向其擴散,需要根據構成碳材料a的碳材料的微孔的表面積在該碳材料的總表面積中所佔的比例是否處於支配地位來改變碳材料a應該滿足的條件。
(微孔的表面積在總表面積中所佔的比例處於支配地位的碳材料a的情況)
在使用微孔的表面積在總表面積中所佔的比例處於支配地位的碳材料a的情況下,由於該微孔變淺,因而對於擔載在碳材料a上的催化劑成分,可以使電解質水溶液與之接觸,而且可以使空氣高效地向其擴散。因此,優選全部滿足以下的條件:即碳材料a的立體結構不發達、碳材料a在總表面積中所佔的微孔的表面積處於支配地位、碳材料a的平均粒徑較小、以及碳材料a的粒徑分布是尖銳的這些條件。此外,「在總表面積中所佔的微孔的表面積處於支配地位」的碳材料a是這樣一種碳材料,它採用氮吸附等溫線的t圖分析得到的微孔表面積smicro與總表面積stotal之比smicro/stotal為0.9以上,其中氮吸附等溫線是採用bet比表面積評價法(brunauer-emmett-teller比表面積評價法)得到的。
在此,所謂bet比表面積評價法,示出了氮氣在液氮溫度下的氮吸附測定評價法,作為直徑2nm以下的孔進行定義的微孔的比表面積smicro、以及總表面積stotal使用由採用bet表面積評價法得到的氮等溫吸附線的t圖分析(日本化學會編コロイド化學i(株)東京化學同人1995年發行)算出的值。
碳材料a的smicro/stotal為0.9以上意味著碳材料a的立體結構不發達(即較多的親水性碳材料粒子以一次粒子的形式存在),且碳材料a在總表面積中所佔的微孔的表面積處於支配地位。在碳材料a的立體結構不發達的情況下,碳材料a的粒子彼此之間緊密配置。因此,碳材料a的體積密度升高。另一方面,在碳材料a的smicro/stotal低於0.9的情況下,碳材料a具有發達的立體結構。也就是說,在此情況下,碳材料a具有多個一次粒子結合而成的複雜的立體結構,在該立體結構的表面擔載著多個催化劑成分。因此,碳材料a成為體積非常大的結構。也就是說,體積密度降低。因此,當在相同的比表面積下對由立體結構不發達的碳材料a構成的催化劑層、和由立體結構發達的碳材料a構成的催化劑層進行比較時,由立體結構不發達的碳材料a構成的催化劑層的層厚減薄。因此,由於空氣的擴散路徑較短,因而可以有效地進行空氣的擴散。其結果是,例如大電流密度放電時的空氣擴散阻力減少,因而大電流密度放電時的電池特性得以改善。
在此,從儘可能增加存在於三相界面的催化劑的角度考慮,需要使儘可能多的空氣和電解液與微孔內的催化劑接觸。但是,如果微孔過深,則空氣和電解液難以達到配置於微孔深部的催化劑。因此,如果微孔過深,則成為效率低下的金屬空氣電池。於是,碳材料a的平均粒徑優選較小。具體地說,碳材料a的平均粒徑優選為低於1.5μm。在此,碳材料a的粒徑是將碳材料a看作為球時的直徑,平均粒徑是d50、即粒徑分布的累積值(所謂的累計分布)為50%時的粒徑。如果碳材料a的平均粒徑較小,則微孔不會極端地深。因此,空氣和電解液容易侵入微孔內。
再者,當使用微孔的表面積在總表面積中所佔的比例處於支配地位的碳材料a時,該碳材料a的粒徑分布是尖銳的。如果粒徑分布過寬,則粒徑較小的親水性碳材料有可能堵塞催化劑層(特別是凝聚相x)。在此情況下,空氣難以擴散至催化劑層內。其結果是,大電流密度放電時空氣擴散阻力增大。因此,電池特性、特別是大電流密度放電時的電池特性降低。於是,在碳材料a中,d90和d10的差(d90-d10)與d50之比(d90-d10)/d50優選低於1.0。在此,d10是採用後述的方法測得的粒徑分布的累積值從較小者開始至10%時的粒徑,d90是該累積值從較小者開始至90%時的粒徑。在此情況下,碳材料a由於幾乎不含有粒徑極端地小的粒子,因而可以抑制催化劑層的堵塞。此外,碳材料a的粒徑分布的測定方法並沒有特別的限制。作為粒徑分布的測定方法,例如有從採用sem(掃描型電子顯微鏡)或tem(透射型電子顯微鏡)取得的圖像中進行觀察、測定的方法、重力沉降法、雷射衍射法、動態光散射法等,可以根據測定對象的碳材料a的特性進行選擇。
此外,當為微孔的表面積在總表面積中所佔的比例處於支配地位的碳材料a時,作為本發明所使用的優選的碳材料,在前述的物質中,特別優選活性炭。活性炭的立體結構不發達,且微孔往往發達,因而smicro/stotal容易達到0.90以上。
(在總表面積中所佔的微孔的表面積未處於支配地位的碳材料a的情況)
另一方面,當使用在總表面積中所佔的微孔的表面積未處於支配地位的碳材料a時,通過使催化劑成分擔載於碳材料a的表面,便對於擔載在碳材料a上的催化劑成分,可以使電解質水溶液與之接觸,而且可以使空氣高效地向其擴散。此外,「在總表面積中所佔的微孔的表面積未處於支配地位」的碳材料a是:採用氮吸附等溫線的t圖分析得到的微孔表面積smicro與總表面積stotal之比smicro/stotal為0.9以下,其中氮吸附等溫線是採用bet比表面積評價法(brunauer-emmett-teller比表面積評價法)得到的。
如果電解質水溶液滲入催化劑層中,則電解質水溶液也侵入碳材料a的微孔中,從而空氣往往難以擴散至微孔內存在的催化劑成分。實際上,由於可以設想存在相當量的在電解質水溶液中擴散而進行反應的氧,因而可以推定在微孔的比較靠近入口的地方存在的催化劑成分將發揮作用。但是,如果微孔所佔的比例極端地升高,則在碳材料a的立體結構以及粒徑分布等未滿足前述條件的情況下,實質上不能發揮作用。在這樣的情況下,例如在微孔的深處存在、氧實質上不能擴散到而無助於電極反應的催化劑成分的比例增加,與擔載的催化劑量相比,恐怕不能得到可以令人滿意的電池性能。
與此相對照,如果選擇微孔表面積smicro與總表面積stotal之比smicro/stotal在0.9以下的碳材料a,則可以抑制實質上無助於電極反應的催化劑成分的比例。如果比smicro/stotal超過0.9,則實質上不進行電極反應的催化劑成分的比例升高,從而難以獲得相對於在電極中使用的催化劑量、經濟上可以令人滿意的電池性能。雖然在理論上可以設想選擇比smicro/stotal接近於0的碳材料a而使實質上不會發揮作用的催化劑成分完全消失的方法,但這樣的比smicro/stotal實質上接近於0的碳材料、即難以利用上述的t圖分析進行微孔的分析的碳材料具有使表面積變得過小的傾向,如果欲高密度擔載催化劑成分,則催化劑成分的粒徑過於增大,在碳材料a上凝聚而成為不均勻的擔載狀態,從而具有不能獲得滿意的電池性能的傾向。如果更優選選擇比smicro/stotal低於0.8且在0.05以上的碳材料a,則無助於電極反應的催化劑成分的影響在電池性能上幾乎不會表現出來,且容易以微細分散的狀態擔載催化劑成分,從而可以得到高效的電池,其能夠獲得以使用的催化劑成分量可以期待的電池性能。
當為在總表面積中所佔的微孔的表面積未處於支配地位的碳材料a時,如果該碳材料a具有某種程度發達的立體結構,則碳材料的細孔部分以外的表面積即外表面積具有增加的傾向。因此,空氣中的氧和電解質水溶液中的oh-離子容易擴散至碳材料的表面上的催化劑成分,即便較少的催化劑量,也可以期待表現出高的電池性能,因而是優選的。這樣的立體結構的發達程度也有採用電子顯微鏡進行觀察來決定的方法,但可以利用dbp吸油量和比表面積的關係進行判斷。
此外,所謂dbp吸油量,是指使鄰苯二甲酸二丁酯與單位質量的碳材料接觸時,被碳材料吸收的鄰苯二甲酸二丁酯的量。鄰苯二甲酸二丁酯(以下簡稱為「dbp」)由於主要被一次粒子的間隙所吸收,因而如果立體結構發達,則dbp吸油量增大,如果立體結構不太發達,則dbp吸油量具有減小的傾向。但是,dbp由於除了一次粒子的間隙以外,也被形成於一次粒子內部的微細孔所吸收,因而dbp吸油量未必直接表現立體結構的發達程度。這是因為如果用氮吸附量測定的比表面積增大,則被微細孔所吸收的dbp增多,從而整個dbp吸油量也具有增大的傾向。因此,具有高立體結構的碳材料在氮吸附量的比例中,dbp吸油量增大。也就是說,即使氮吸附量較小,dbp吸油量也增大。相反,立體結構不發達的碳材料在氮吸附量的比例中,dbp吸油量減小。也就是說,氮吸附量即使較大,dpb吸油量也減小。
碳材料a如果使用dbp吸油量xml/100g和基於bet比表面積評價的比表面積sbetm2/g之比x/sbet在0.05以上的碳材料,則空氣中的氧和電解質水溶液中的oh-離子容易擴散,從而即使以較少的催化劑量,也可以獲得表現出高電池性能的高性能的催化劑層。比x/sbet之比如果低於0.05,則外表面積的比例降低,空氣中的氧和電解質水溶液中的oh-離子難以擴散,因而較少的催化劑量往往難以穩定地發揮催化劑層的性能。如果超過3.0,則機械強度往往降低,在編入電池中使用時,立體結構發生破壞,從而往往不能獲得期待的電池性能。
[碳材料b]
在本發明所使用的碳材料中,碳材料b通過含在滲入電解質水溶液而發揮作用的催化劑層中,從而形成確保不會滲入電解質水溶液的區域、使含有放電反應所需要的氧的空氣擴散至催化劑層的路徑。因此,碳材料b優選為對電解質水溶液難以潤溼的材料。
作為碳材料b,如果選擇25℃、相對溼度10%下的水蒸氣吸附量低於0.1cm3/g的碳材料,則與電解質水溶液的親和性降低,碳材料b的表面難以被電解質水溶液潤溼,從而使空氣容易存在於碳材料b的附近。如果選擇0.1cm3/g以上的碳材料作為碳材料b,則放電反應大多不會高效地進行。可以推定這是因為:與電解質水溶液的親和性升高,碳材料b部分地被電解質水溶液潤溼,從而催化劑層中的空氣擴散路徑變得貧乏。
與此相對照,如果選擇25℃、相對溼度10%下的水蒸氣吸附量低於0.05cm3/g的碳材料b,則在碳材料b的周圍可以更切實地存在空氣,從而可以獲得較高的電池性能。碳材料b一般認為理想的也是實質上與電解質水溶液幾乎不具有親和性的碳材料、即25℃、相對溼度10%下的水蒸氣吸附量幾乎為0的材料。
作為相對溼度10%下的水蒸氣吸附量幾乎為0的材料,可以設想立體結構幾乎不發達的碳材料、幾乎不具有有助於與電解質水溶液的親和性的表面官能團和微細的表面凹凸結構的碳材料、以及例如經過耗費非常高的成本的石墨化工序而製作的碳材料等。但是,在使用立體結構幾乎不發達的碳材料的金屬空氣電池中,電池性能不會如所期待的那樣升高。另外,可以設想使用經過耗費非常高的成本的石墨化工序而製作的碳材料的金屬空氣電池大多在經濟上是不相稱的。從這樣的角度考慮,25℃、相對溼度10%下的水蒸氣吸附量的下限可以設想為0.01cm3/g左右。
再者,如果碳材料b具有發達的立體結構,則可以在其結構內積聚空氣,因而是優選的。特別在凝聚相y中,通過使碳材料b彼此之間凝聚,便有可能使立體結構中的空間以三維的方式連結起來,從而能夠以比將單一粒子分散於催化劑層中的情況更少的量,使空氣容易向催化劑層內擴散的粗的路徑變得發達。另外,在凝聚相x的內部也以單一粒子的形式微細分散的碳材料b的立體結構中的空間可以作為微細的空氣擴散路徑發揮作用。
作為碳材料b優選的材料的例子,可以使用碳黑、石墨、碳纖維、活性炭等或它們的粉碎物、碳納米纖維、碳納米管、石墨烯等的碳化合物等。另外,使用各種鑄型而形成了結構的碳材料也可以使用。也可以混合使用上述的2種以上。另外,也可以改變分散於凝聚相x中的碳材料b和作為凝聚相y的主成分的碳材料b的種類和組成而使用。碳材料b特別優選的材料的例子為碳黑。碳黑是多個一次粒子熔合在一起而形成被稱之為「結構(structure)」的立體結構。根據種類的不同,該結構發達而成為一次粒子的連結環抱空間的結構。如果使用具有這樣的結構的碳材料b,則環抱的空間成為氣體的擴散路徑,因而是優選的。立體結構的發達程度也有採用電子顯微鏡進行觀察來決定的方法,但可以利用dbp吸油量和比表面積的關係進行判斷。
碳材料b如果使用dbp吸油量xml/100g與基於bet比表面積評價的比表面積sbetm2/g之比x/sbet在0.5以上的碳材料,則可以得到氣體擴散路徑容易確保的高性能的催化劑層。如果比x/sbet之比低於0.5,則作為氣體擴散路徑的空間變得貧乏,從而往往難以穩定地發揮催化劑層的性能。如果超過3.0,則機械強度往往降低,在編入電池中使用時,立體結構發生破壞,從而往往不能獲得期待的電池性能。特別地,如果x/sbet在1.0以上,則碳材料所環抱的空間充分地增大,可以切實地形成粗的氣體擴散路徑,因而可以獲得穩定的電池性能。
本發明的催化劑層中的碳材料b優選的含有率受到碳材料a的種類、凝聚相x與凝聚相y的分配率、催化劑成分的種類、相對於碳材料a的擔載率以及粒徑等的影響。因此,如前所述,碳材料b優選的含有率也隨著構成碳材料a的碳材料的微孔的表面積在該碳材料的總表面積中所佔的比例是否處於支配地位而稍有變動。
(1)當使用微孔的表面積在總表面積中所佔的比例處於支配地位的碳材料a時,碳材料b在催化劑層中的含有率優選為超過10質量%且低於50質量%的範圍。如果在該範圍外,則本發明提出的催化劑層結構往往不能形成,從而不會成為效率良好的金屬空氣電池。例如如果在10質量%以下,則碳材料b往往難以形成對空氣擴散有效的凝聚相y。另外,例如如果在50質量%以上,則含有催化劑成分的凝聚相x的一部分往往成為不取連續結構而分散於凝聚相y中的結構。此外,更優選的例示的範圍超過10質量%且在40質量%以下。如果例示出進一步優選的範圍,則為30質量%~35質量%。
(2)與此相對照,當使用在總表面積中所佔的微孔的表面積未處於支配地位的碳材料a時,優選為5質量%以上且低於50質量%的範圍。如果在該範圍外,則本發明提出的催化劑層結構往往不能形成,從而不會成為效率良好的金屬空氣電池。例如如果低於5質量%,則碳材料b往往難以形成對空氣擴散有效的凝聚相y。另外,例如如果在50質量%以上,則含有催化劑成分的凝聚相x的一部分往往成為不取連續結構而分散於凝聚相y中的結構。此外,如果例示出更優選的範圍,則為10質量%~40質量%。
本發明中含有的碳材料a和碳材料b對電解質水溶液的潤溼性的控制、即25℃、相對溼度10%下的水蒸氣吸附量的控制可以通過從通常存在的碳材料中以水蒸氣吸附量為指標進行選擇而實現。或者,即使在為具有小於優選範圍的水蒸氣吸附量的碳材料的情況下,也可以通過對碳材料用酸或鹼等處理碳材料表面、或者曝露於氧化氣氛環境下,使水蒸氣吸附量增加到優選的範圍。雖然並沒有進行限定,但可以採用如下的方法增加水蒸氣吸附量:例如在加熱的濃硝酸中進行處理,浸漬於雙氧水溶液中,在氨氣流中進行熱處理,浸漬於加熱的氫氧化鈉水溶液中,在koh或naoh中進行加熱,或者在稀薄氧、稀薄no或no2中進行加熱處理。相反,在水蒸氣吸附量過多的情況下,通過在不活潑氣氛下進行燒成,也可以將水蒸氣吸附量降低至優選的範圍。雖然並沒有進行限定,但例如通過在氬、氮、氦、真空等氣氛下進行加熱處理,可以使水蒸氣吸附量降低。
[空氣極的催化劑層的製作方法]
在本發明的金屬空氣電池的空氣極的催化劑層的製作方法中,只要能夠以凝聚相y分散於凝聚相x的連續體中的方式進行製作,就沒有特別的限定。能夠在以擔載著催化劑成分的碳材料a和碳材料b為主成分的材料中,根據需要添加水或有機溶劑而製作出油墨,將該油墨乾燥成膜狀,從而形成催化劑層。
特別優選的催化劑層的製作方法如下所述。
(1)使用微孔的表面積在總表面積中所佔的比例處於支配地位的碳材料a的情況
[優選的催化劑層的製作方法1-1]
(i)首先,通過將碳材料a粉碎,從而將碳材料a的平均粒徑設定為低於1.5μm。在此,將碳材料a粉碎的方法並沒有特別的限制。作為碳材料a的粉碎方法,例如可以列舉出利用超聲波的方法、使用球磨機或玻璃珠等而進行機械粉碎的方法等。
(ii)接著,調整碳材料a的粒徑分布,以便使碳材料a的粒徑分布變得尖銳。具體地說,調整碳材料a的粒徑分布,以便滿足(d90-d10)/d50<1.0的條件。在此,對碳材料a的粒徑分布進行調整的方法並沒有特別的限制。作為調整碳材料a的粒徑分布的方法,例如可以列舉出使用篩子的篩分、使用風力分級機的方法等。優選的方法是使用風力分級機的方法。
(iii)接著,使催化劑成分擔載在碳材料a上。
(iv)接著,將擔載著催化劑成分的碳材料a和碳材料b在溶劑中進行粉碎混合而製作出油墨,使其油墨乾燥,對得到的固體物質進行乾式粉碎,從而製作出凝聚相x前體。
(v)接著,將在溶劑中粉碎混合碳材料b而製作的油墨乾燥,對得到的固體物質進行乾式粉碎,從而製作出凝聚相y前體。在溶劑中對得到的凝聚相x前體和凝聚相y前體進行攪拌混合,將製作的油墨乾燥成膜狀,從而設計為催化劑層。
在該方法中,在凝聚相x前體和凝聚相y前體中不含有作為粘結劑的成分,固體成分粒子實質上僅通過範德瓦爾斯力粘著在一起。因此,在乾式粉碎或攪拌混合時,必須調整強度或時間,以便使凝聚結構不會變得過細。具體的乾式粉碎和攪拌混合的程度由於隨使用的材料和量、器具的不同而不同,因而不能進行限定,但可以調整乾式粉碎和攪拌混合的強度或時間,以便在得到的催化劑層斷面的面積為10μm×10μm的視場中,當量圓直徑在300nm以上這種大小的沒有催化劑成分的碳材料凝聚相(凝聚相y)分散1個以上。此外,面積為10μm×10μm的視場中的凝聚相y的個數的上限值並沒有特別的限制,只要凝聚相x的連續結構不會遭到破壞即可。
[優選的催化劑層的製作方法1-2]
(i)首先,與所述優選的催化劑層的製作方法1同樣,對碳材料a的平均粒徑和粒徑分布進行調整。
(ii)接著,使催化劑成分擔載在碳材料a上。
(iii)接著,在溶劑中將擔載著催化劑成分的碳材料a、碳材料b以及作為粘結劑的聚四氟乙烯(ptfe)粉碎混合,從而製作出油墨,將其油墨乾燥成膜狀,進而根據需要進行熱處理或者熱壓,通過粘結劑而使碳材料a和碳材料b熱熔合在一起,對得到的固體物質進行乾式粉碎,從而製作出凝聚相x前體。
(iii)接著,在溶劑中將碳材料b和作為粘結劑的ptfe粉碎混合,從而製作出油墨,將其油墨乾燥成膜狀,進而根據需要進行熱處理或者熱壓,通過粘結劑而使碳材料b熱熔合在一起,對得到的固體物質進行乾式粉碎,從而製作出凝聚相y前體。
(v)在溶劑中對得到的凝聚相x前體和凝聚相y前體進行攪拌混合,將製作的油墨乾燥成膜狀,從而設計為催化劑層。
如果為該方法,則凝聚結構的機械強度升高,在催化劑層的形成途中可以防止凝聚相的破壞,從而容易得到如目標那樣的結構,因而是優選的。含有粘結劑的凝聚相無論是只有凝聚相x還是只有凝聚相y都沒關係。具體的乾式粉碎和攪拌混合的程度由於隨使用的材料和量、器具的不同而不同,因而不能進行限定,但可以調整乾式粉碎和攪拌混合的強度或時間,以便在得到的催化劑層斷面的面積為10μm×10μm的視場中,當量圓直徑在300nm以上這種大小的沒有催化劑成分的碳材料凝聚相(凝聚相y)分散1個以上。
(2)使用在總表面積中所佔的微孔的表面積未處於支配地位的碳材料a的情況
[優選的催化劑層的製作方法2-1]
(i)將擔載著催化劑成分的碳材料a和碳材料b在溶劑中進行粉碎混合而製作出油墨,使該油墨乾燥,對得到的固體物質進行乾式粉碎,從而製作出凝聚相x前體。
(ii)接著,將在溶劑中粉碎混合碳材料b而製作的油墨乾燥,對得到的固體物質進行乾式粉碎,從而製作出凝聚相y前體。在溶劑中對得到的凝聚相x前體和凝聚相y前體進行攪拌混合,將製作的油墨乾燥成膜狀,從而設計為催化劑層。
在該方法中,在凝聚相x前體和凝聚相y前體中不含有作為粘結劑的成分,固體成分粒子實質上僅通過範德瓦爾斯力粘著在一起。因此,在乾式粉碎或攪拌混合時,必須調整強度或時間,以便使凝聚結構不會變得過細。具體的乾式粉碎和攪拌混合的程度由於隨使用的材料和量、器具的不同而不同,因而不能進行限定,但可以調整乾式粉碎和攪拌混合的強度或時間,以便在得到的催化劑層斷面的面積為10μm×10μm的視場中,當量圓直徑在300nm以上這種大小的沒有催化劑成分的碳材料凝聚相(凝聚相y)分散1個以上。此外,面積為10μm×10μm的視場中的凝聚相y的個數的上限值並沒有特別的限制,只要凝聚相x的連續結構不會遭到破壞即可。
[優選的催化劑層的製作方法2-2]
(i)首先,在溶劑中將擔載著催化劑成分的碳材料a、碳材料b以及作為粘結劑的ptfe粉碎混合,從而製作出油墨,將其油墨乾燥成膜狀,進而根據需要進行熱處理或者熱壓,通過粘結劑而使碳材料a和碳材料b熱熔合在一起,對得到的固體物質進行乾式粉碎,從而製作出凝聚相x前體。
(ii)接著,在溶劑中將碳材料b和作為粘結劑的ptfe粉碎混合,從而製作出油墨,將其油墨乾燥成膜狀,進而根據需要進行熱處理或者熱壓,通過粘結劑而使碳材料b熱熔合在一起,對得到的固體物質進行乾式粉碎,從而製作出凝聚相y前體。在溶劑中對得到的凝聚相x前體和凝聚相y前體進行攪拌混合,將製作的油墨乾燥成膜狀,從而設計為催化劑層。
如果為該方法,則凝聚結構的機械強度升高,在催化劑層的形成途中可以防止凝聚相的破壞,從而容易得到如目標那樣的結構,因而是優選的。含有粘結劑的凝聚相無論是只有凝聚相x還是只有凝聚相y都沒關係。具體的乾式粉碎和攪拌混合的程度由於隨使用的材料和量、器具的不同而不同,因而不能進行限定,但可以調整乾式粉碎和攪拌混合的強度或時間,以便在得到的催化劑層斷面的面積為10μm×10μm的視場中,當量圓直徑在300nm以上這種大小的沒有催化劑成分的碳材料凝聚相(凝聚相y)分散1個以上。
[正極的電極結構]
本發明的金屬空氣電池的正極的電極結構只要是在負極對置側配置有催化劑層、來自相反側的大氣中的空氣可以擴散、且從負極對置側滲入催化劑層中的電解質水溶液不會向大氣開放側漏出的結構,且賦予適當的集電結構,就沒有特別的限定。這樣的電極結構不論所使用的碳材料a的微孔的表面積在該碳材料的總表面積中所佔的比例是否處於支配地位,都可以適用於本發明的金屬空氣電池的正極的電極結構。
一般地說,優選的是在負極對置側配置本發明的催化劑層、接著為用疏水性多孔質材料形成的氣體擴散層這兩層結構。此時,集電體只要與兩層結構的至少一部分接觸即可,優選的是可以配置在催化劑層和氣體擴散層之間、或者氣體擴散層的大氣開放側。集電體並不優選在與鹼水溶液接觸的環境下發生溶解、或者電子傳導性發生變化的材料,而優選化學性質穩定且與碳材料的接觸電阻較小的材料。一般地說,為鎳網、或實施了鎳鍍覆的不鏽鋼網。
在比氣體擴散層更靠大氣開放側使用集電體的情況下,氣體擴散層必須由在催化劑層和集電體之間能夠進行電子傳導的具有電子傳導性的材料構成。氣體擴散層要求具有在放電時從大氣開放側吸取空氣、在氣體擴散層中擴散、並使空氣氣體均勻地擴散至催化劑層的功能,以及在催化劑層和集電體之間傳導電子的功能,只要最低限度地具有這些功能,就並沒有特別的限定。在通常的例子中,碳布和碳紙等碳材料優選為主要的構成材料。除氣體擴散性、電子傳導性以外,只要還能夠賦予耐蝕性,也可以使用金屬網和金屬毛絨等金屬材料。作為進一步優選的氣體擴散層的結構的例子,可以列舉出2層結構:其由氣體擴散層的大氣開放側為以纖維狀碳材料為主成分的氣體擴散纖維層、催化劑層側的層為以難於被水潤溼的碳黑為主成分的微孔層構成。
在形成催化劑層時,作為將含有催化劑成分、碳材料a以及碳材料b為主成分的油墨乾燥成膜狀的方法,可以適用通常提出的方法,並沒有特別的限定。例如,如果在氣體擴散層上塗布油墨,則可以列舉出刷塗、噴塗、輥塗、噴墨、絲網印刷等方法。或者也可以選擇如下的方法:採用棒塗、刷塗、噴塗、輥塗、噴墨、絲網印刷等方法塗布油墨並使其乾燥,在ptfe片材或ptfe板等高分子材料的表面上,暫且在其它材料上形成催化劑層後,採用熱壓等方法與氣體擴散層或隔膜接合在一起。
實施例
[實施例1a~31a以及比較例1a~21a]
<碳材料a的準備以及物性的測定>
在示出本發明的金屬空氣電池用電極的實施例1a~31a時,作為碳材料a,準備10種碳材料a1~j1。表1(碳材料的種類和其物性)示出了各種碳材料的各種物性。
此外,碳材料a1是沒有進行平均粒徑和粒徑分布的調整的碳材料,是作為比較例使用的。碳材料b1是進行過平均粒徑和粒徑分布的調整的碳材料。碳材料c1是進行過平均粒徑和粒徑分布的調整的碳材料。碳材料c1的平均粒徑較小,粒徑分布也是尖銳的。在本實施例的評價對象中為最優良品。碳材料d1是進行過平均粒徑和粒徑分布的調整的碳材料。碳材料d1的水蒸氣吸附量稍低。碳材料e1是進行過平均粒徑和粒徑分布的調整的碳材料。碳材料e1的氮吸附比表面積稍大。碳材料f1是沒有進行平均粒徑的調整而進行過粒徑分布的調整的碳材料。碳材料f1的粒徑分布是尖銳的,但平均粒徑為1.5μm以上。因此,碳材料f1作為比較例使用。碳材料h1是進行過平均粒徑的調整,但沒有進行粒徑分布的調整的碳材料。碳材料h1的平均粒徑低於1.5μm,但粒徑分布是寬的。因此,碳材料h1作為比較例使用。以上的碳材料a1~h1為活性炭。碳材料i1、j1為碳黑,相對於總表面積的微孔的表面積未處於支配地位。因此,碳材料i1、j1作為比較例使用。
在此,碳材料的平均粒徑的調整通過將碳材料粉碎來進行。碳材料的粉碎使用フリッチェ公司生產的行星球磨機p-7。另外,碳材料的粒徑分布的調整通過對碳材料進行分級來進行。碳材料的分級通過調整日鉄鉱業株式公司生產的肘射流分級機(elbowjetclassifier)的分級邊緣位置來進行。粒徑分布的測定採用雷射衍射法來進行。
另外,關於氮吸附比表面積sbet,使用自動比表面積測定裝置(日本ベル生產,belsorp36)並採用氮氣對在120℃下真空乾燥過的樣品進行測定,利用基於bet法的1點法來決定比表面積sbet。另外,t圖分析使用裝置附帶的分析程序算出總表面積stotal以及微孔表面積smicro。水蒸氣吸附量使用定容量式水蒸氣吸附裝置(日本ベル生產,belsorp18)進行測定,將在120℃、1pa以下進行過2小時脫氣前處理的試料保持在25℃的恆溫中,在從真空狀態至25℃下的水蒸氣的飽和蒸氣壓之間,慢慢地供給水蒸氣而使相對溼度階段性地發生變化,從而測定水蒸氣吸附量。由得到的測定結果描繪出吸附等溫線,從圖中讀取相對溼度10%時的水蒸氣吸附量。在表1中,將讀取的水蒸汽量換算成每1g試料所吸附的標準狀態的水蒸氣體積並示出。
<碳材料b的準備以及物性的測定>
在示出本發明的金屬空氣電池用電極的實施例1a~31a時,作為碳材料b,準備6種碳材料k1~p1。表2(碳材料的種類和其物性)示出了各種碳材料的各種物性。在此,碳材料k1是立體結構不發達,水蒸氣吸附量也大的碳材料。碳材料k1作為比較例使用。碳材料l1是立體結構發達,但水蒸氣吸附量較大的碳材料。碳材料l1也作為比較例使用。碳材料m1是立體結構某種程度(滿足本實施方式的條件的程度)地發達的碳材料,水蒸氣吸附量也小的碳材料。碳材料n1是立體結構比碳材料m1更發達的碳材料,水蒸氣吸附量也小。碳材料o1是立體結構比碳材料n1更發達的碳材料,水蒸氣吸附量也小。碳材料p1的立體結構在碳材料k1~o1中最為發達,水蒸氣吸附量也最低。碳材料p1在本實施例的評價對象中為最優良品。
此外,碳材料k1~p1的氮吸附比表面積sbet、總表面積stotal、微孔表面積smicro、水蒸氣吸附量是採用與碳材料a1~j1同樣的方法測得的值。dbp吸油量x(cm3/100g)使用吸收儀(absorptiontometer,brabender公司生產),將最大轉矩的70%時的dbp添加量換算成每100g試料的dbp吸油量來決定。
<催化劑的調配>
將擔載著催化劑的碳材料作為碳材料a,從表1的碳材料中選擇1種作為碳材料a,採用以下的方法調配擔載著pt的催化劑作為催化劑成分。在氯鉑酸水溶液中,分散作為碳材料a的從表1的碳材料中選擇的1種,然後在50℃下保溫,並一邊攪拌一邊添加雙氧水,接著添加na2s2o4水溶液,從而獲得催化劑前體。在對該催化劑前體進行過濾、水洗、乾燥後,在100%h2氣流中,於300℃下進行3小時的還原處理,從而調配出在催化劑載體碳材料上擔載著40質量%pt的pt催化劑。
在製作本發明的金屬空氣電池用電極時,製作了以下的塗布料漿。
<催化劑層油墨的製作法1>
在乙醇中添加作為起始原料的按上述那樣製作的40質量%的催化劑、以及根據需要添加從表1中選擇的沒有擔載催化劑成分的碳材料、作為粘結劑的ptfe(ダイキン工業生產,ptfe分散液,d-210c),採用直徑1mm的玻璃珠將其粉碎後,除去玻璃珠,利用乙醇進行濃度調整,以便使料漿中的鉑濃度為0.25質量%,從而製作出不具有凝聚結構的催化劑層油墨。
<催化劑層油墨的製作法2>
在乙醇中添加作為起始原料的按上述那樣製作的40質量%的催化劑、以及根據需要添加作為碳材料b的從表1中選擇的沒有擔載催化劑成分的碳材料、作為粘結劑的ptfe(ダイキン工業生產,ptfe分散液,d-210c),採用直徑1mm的玻璃珠將其粉碎後,除去玻璃珠,對得到的料漿進行真空乾燥,從而得到碳材料a凝聚物。接著,在乙醇中添加作為起始原料的從表1中選擇的沒有擔載催化劑成分的碳材料b、以及根據需要添加作為粘結劑的ptfe(ダイキン工業生產,ptfe分散液,d-210c),採用直徑1mm的玻璃珠將其粉碎後,除去玻璃珠,對得到的料漿進行真空乾燥,從而得到碳材料b凝聚物。在此,當在碳材料a凝聚物、或者碳材料b凝聚物中含有粘結劑時,在氬氣流中於320℃下對得到的凝聚物進行熱處理,從而進行熔合處理。
接著,在乙醇中添加得到的碳材料a凝聚物和碳材料b凝聚物,採用直徑1mm的玻璃珠對其進行粉碎和攪拌處理,利用乙醇進行濃度調整,以便使料漿中的pt濃度為0.25質量%,從而形成具有凝聚結構的催化劑層油墨。此外,關於粉碎和攪拌處理的強度,可事先進行調整和決定,以便在使用按實施例1a的組成製作的催化劑層油墨而形成的催化劑層斷面的面積為10μm×10μm的視場中,使當量圓直徑在300nm以上這種大小的沒有催化劑成分的碳材料凝聚相(凝聚相y)分散1個以上,然後將其條件適用於所有的情況。
<電極形成>
作為疏水性多孔質層,使用由微孔層(mpl)層疊而成的碳紙(sglカーボン生產的gdl24bc)。將疏水性多孔質層裁切成10cm見方(100cm2),採用噴塗對按上述那樣製作的pt濃度為0.25質量%的催化劑層油墨進行再塗(recoat),並在90℃下進行真空乾燥,從而製作出空氣電池用正極。此外,在此,在催化劑層油墨的製作法1的方法含有粘結劑的情況下,在氬氣流中於320℃下對得到的電極進行熱處理,從而進行熔合處理。
另外,測量塗布前後的疏水性多孔質層的質量變化,算出pt單位面積重量,對塗布量進行調整,從而使鉑單位面積重量為0.20mg/cm2。
<金屬空氣電池性能評價用硬幣電池的製作>
為了對得到的電極進行評價,製作了硬幣電池。在兼作負極端子的內徑為20mm的硬幣電池用殼體上以沒有凹凸的方式均勻地塗敷0.30g的zn粉末(高純度化學社生產,粒徑為75μm),從而形成負極。接著,在塗敷的zn粉末上滴加140μl的8mol/l的koh水溶液而使其滲入,從其上放置直徑為20mm的膜濾器(アドバンテック生產的親水性ptfeh100)作為隔膜,也使8mol/l的koh水溶液滲入隔膜中。再者,採用直徑14mm的衝頭對按上述那樣製作的空氣電池用電極進行衝裁,使催化劑層塗布側成為下側而重疊在隔膜上。再者,在其上鋪上作為正極集電體的直徑19mm的鎳網,並在作為正極端子的硬幣電池用帽蓋中開4個內徑為2mm的空氣孔,進而安裝短路防止用墊片,將其蓋上,便形成評價用硬幣電池。
<性能評價>
評價用硬幣電池在製作後,迅速地進行了性能評價。在評價中,將製作的評價用硬幣電池的下側殼體設定為負極,將上側帽蓋設定為正極,用能夠進行壓力控制的氣缸(cylinder)狀端子夾持硬幣電池,將氣缸端子的壓力控制為75kg/cm2,以便不會堵塞在帽蓋上形成的空氣孔,然後在室溫下以100ma的恆電流進行放電,將從放電開始10分鐘後的單電池電壓記錄為電池性能。這樣一來,在本實施例中,能夠以較高的電流進行放電。此時的單電池電壓依賴於催化劑層的空氣擴散阻力而變動。也就是說,空氣擴散阻力越低,單電池電壓越具有增大的傾向。
<性能評價結果1>
表3示出了使用由作為比較例的催化劑層油墨的製作法1調配的催化劑層油墨而形成的電極的組成、和編入了各自的電極的空氣電池的性能評價結果,表4示出了使用由催化劑層油墨的製作法2調配的催化劑層油墨而形成的電極的組成、和編入了各自的電極的空氣電池的性能評價結果。
此外,在表3和表4所示的所有情況下,擔載著催化劑的碳材料a使用全部滿足本發明的要件的碳材料b1,作為催化劑成分的pt的單位面積重量統一為0.20mg/cm2。另外,在使用未擔載催化劑的碳材料b的情況下,將種類統一為滿足作為本發明要件的水蒸氣吸附量和比x/sbet的碳材料o1,且催化劑層中的含量也統一為合計30質量%。另外,即使在含有粘結劑的情況下,種類統一為ptfe,催化劑層中的含有率統一為合計10wt%。
表4所示的實施例1a~4a無論在哪一種情況下,與表3以及表4所示的比較例1a~8a相比,顯示出更高的單電池電壓,顯示出更優良的電池特性。特別地,由沒有凝聚結構的方法製作的比較例1a~4a無論在哪一種情況下,單電池電壓不穩定,從放電開始電壓持續下降,10分鐘後低於0.6v。另外,對以具有凝聚結構的方式製作的比較例5a~8a、以及實施例1a~4a進行比較,凝聚相x中包含不具有與水的潤溼性的碳材料o1的實施例1a~4a相對於凝聚相x中不包含不具有與水的潤溼性的碳材料o1的比較例5a~8a,顯示出較高的單電池電壓,具有優良的電池性能。如果在實施例1a~4a中進行比較,則完全不含有粘結劑的實施例1a在其中性能最差,僅在凝聚相y中含有粘結劑的實施例3a具有最優良的性能。
接著,就表3所示的比較例1a~4a的催化劑層、以及表4所示的實施例1a~4a的催化劑層進行了斷面結構觀察。對於觀察試料,用刀片以10mm左右見方的大小切出製作的電極,在用環氧進行樹脂填埋後,固定在冷凍切片機的夾具上,以便能夠切削催化劑層的斷面。將製作的夾具設置在切片機上,刀設置金剛石修整刀。此時,使金剛石修整刀相對於刀的行走方向偏移10度左右的角度,從而使催化劑層被傾斜地切削。
在修整後,繼續使用金剛石修整刀,沿催化劑層的深度方向以每1次50nm的速度至少切削100次,從而製作出催化劑層的切斷面。將製作了這些切斷面的催化劑層設置在電子顯微鏡上,以1萬倍的放大倍數對2次電子像和背散射電子圖像進行了觀察。在表3所示的比較例1a~4a的催化劑層中,背散射電子圖像以均勻明亮的對比度而被觀察到,沒有擔載催化劑成分的碳材料的凝聚相(氣體擴散碳材料凝聚相)沒有看到。與此相對照,表4所示的實施例1a~4a的催化劑層在由2次電子像可以明顯地判別存在碳材料的部位中,可以觀察到在背散射電子圖像中成為暗的對比度的部位、即沒有擔載催化劑成分的碳材料的凝聚相(氣體擴散碳材料凝聚相)。
為了更定量地進行識別,對於背散射電子圖像,在1萬倍的放大倍數下採用272dpi×272dpi以上的解析度、且以256色的層次的亮度採集作為電子信息,對於採集的圖像的亮度,使用圖像分析軟體進行二值化,從而將從暗的至第110層次的範圍用黑色表示,並使從第111層次向明的直至第256層次的範圍成為白色。接著,對各黑色點進行1次膨脹處理,從而對相鄰的點彼此之間進行識別。再者,執行填孔處理,對範圍內的空白部分進行填孔,從而識別為同一範圍。最後,進行使膨脹的部分復原的收縮處理,從而使目標的範圍明確化。而且由各黑色部分的面積算出各黑色部分的當量圓直徑,並將低於300nm的部分全部切除。在剩餘的黑色部分中,如果在同視場的二次電子像中測量存在碳材料的黑色部分的個數,則在實施例1a~4a的所有情況下為1個以上。
<性能評價結果2>
接著,在改變凝聚相x中含有的碳材料a的種類時進行了性能評價。表5示出了使用由催化劑層油墨的製作法2調配的催化劑層油墨而形成的電極的組成、和編入了各自的電極的空氣電池的性能評價結果。
此外,在表5所示的催化劑層中,作為催化劑成分的pt的單位面積重量統一為0.20mg/cm2,關於構成成分在催化劑層中的含有率,催化劑成分為22質量%,碳材料a為33質量%,碳材料b在凝聚相x中含有的部分為10質量%,在凝聚相y中含有的部分為15質量%,粘結劑不含在凝聚相x中,只含在凝聚相y中,其含有率統一為20質量%。凝聚相x中含有的碳材料b和凝聚相y中含有的碳材料b都使用滿足本發明要件的碳材料n1,粘結劑設定為ptfe。
將完全滿足與本發明的碳材料a有關的要件的碳材料b1、c1、d1、e1、g1用作碳材料a的實施例5a~9a和使用並不滿足與本發明的碳材料a有關的要件的至少1項以上的碳材料a1、f1、h1、i1、j1的比較例9a~13a相比,可以產生更高的單電池電壓,從而顯示出優良的電池性能。特別地,將平均粒徑較小、粒徑分布也尖銳的碳材料c1用作碳材料a的實施例6a的特性最為良好。
<性能評價結果3>
接著,在改變凝聚相x中含有的碳材料b的種類和催化劑層中的含有率時進行了性能評價。表6示出了使用由催化劑層油墨的製作法2調配的催化劑層油墨而形成的電極的組成、和編入了各自的電極的空氣電池的性能評價結果。
此外,在表6所示的催化劑層中,作為催化劑成分的pt的單位面積重量統一為0.20mg/cm2,關於碳材料b在催化劑層中的含有率,凝聚相y中含有的部分為10質量%,粘結劑不含在凝聚相x中,只含在凝聚相y中,統一含有10質量%。碳材料a使用滿足本發明要件的碳材料d1,凝聚相y中含有的碳材料b使用滿足本發明要件的碳材料o1,粘結劑使用ptfe。
碳材料m1、n1、o1、p1滿足與本發明的碳材料b有關的要件即25℃、相對壓力為0.1的環境下的水蒸氣吸附量低於0.1cm3/g,dbp吸油量x(cm3/100g)和基於bet評價的比表面積sbet之比x/sbet為0.5以上這兩者,將該碳材料m1、n1、o1、p1用作在凝聚相x中含有的碳材料b的實施例10a~18a和將並不滿足與本發明的碳材料b有關的要件的碳材料k1、l1用作凝聚相x中含有的碳材料b的比較例14a~15a相比,可以產生更高的單電池電壓,從而顯示出優良的電池性能。
特別地,在將水蒸氣吸附量低於0.05cm3/l的碳材料o1、p1用作碳材料b的實施例15a、18a(關於碳材料o1,著眼於催化劑層中的含量與其它實施例相同的實施例15a)中,其電池特性特別良好。另外,在表6中,如果對使凝聚相x中含有的碳材料b在催化劑層中的含有率於0~40質量%之間發生變化的比較例16a、17a以及實施例12a~17a進行比較,則凝聚相x中不含有碳材料b的比較例16a、凝聚相x中含有40質量%的碳材料b、且凝聚相y中也含有10質量%的碳材料b、從而催化劑層中的碳材料b的合計含有率為50質量%的比較例17a與凝聚相x中和凝聚相y中都含有碳材料b、催化劑層中的合計含有率超過10質量%且低於50質量%的實施例12a~17a相比,為較差的性能。另外,在催化劑層中的合計含有率超過10質量%且在40質量%以下的情況下,其特性變得特別良好。另外,催化劑層中的碳材料b的合計含有率為30質量%的實施例16a在實施例12a~17a的中,特性最為良好。另外,在凝聚相x中的碳材料b的含有率為0.1~0.5的實施例13a~17a中,其特性變得特別良好。
<性能評價結果4>
接著,在改變凝聚相y中含有的碳材料b的種類和催化劑層中的含有率時進行了性能評價。表7示出了使用由催化劑層油墨的製作法2調配的催化劑層油墨而形成的電極的組成、和編入了各自的電極的空氣電池的性能評價結果。
此外,在表7所示的催化劑層中,作為催化劑成分的pt的單位面積重量統一為0.20mg/cm2;關於碳材料b在催化劑層中的含有率,凝聚相x中含有的部分為10質量%;粘結劑不含在凝聚相x中,只含在凝聚相y中,催化劑層中的含有率統一為10質量%。碳材料a使用滿足本發明要件的碳材料c1,凝聚相x中含有的碳材料b使用滿足本發明要件的碳材料p1,粘結劑使用ptfe。
碳材料m1、n1、o1、p1滿足與本發明的碳材料b有關的要件即25℃、相對壓力為0.1的環境下的水蒸氣吸附量低於0.1cm3/g,dbp吸油量x(cm3/100g)和基於bet評價的比表面積sbet之比x/sbet為0.5以上這兩者的條件,將該碳材料m1、n1、o1、p1用作在凝聚相y中含有的碳材料b的實施例21a~29a和將並不滿足與本發明的碳材料b有關的要件的碳材料k1、l1用作凝聚相y中含有的碳材料b的比較例18a、19a相比,可以產生更高的單電池電壓,從而顯示出優良的電池性能。
另外,在表7中,如果對使凝聚相y中含有的碳材料b在催化劑層中的含有率於0~45質量%之間發生變化的比較例20a、21a以及實施例24a~29a進行比較,則凝聚相y中不含有碳材料b的比較例20a、凝聚相x中含有10質量%的碳材料b、且凝聚相y中也含有45質量%的碳材料b、從而催化劑層中的碳材料b的合計含有率為50質量%以上的比較例21a與凝聚相x中和凝聚相y中都含有碳材料b、催化劑層中的合計含有率超過10質量%且低於50質量%的實施例24a~29a相比,為較差的性能。另外,催化劑層中的碳材料b的合計含有率為35質量%的實施例28a在實施例24a~29a中,特性最為良好。
另外,在表7所示的催化劑層中,對於實施例25a和27a的催化劑層,採用與<性能評價結果1>同樣的方法,進行了斷面結構觀察,結果在300nm以上的黑色部分中,如果在同視場的二次電子像中測量存在碳材料的黑色部分的個數,則確認為1個以上。再者,如果削除當量圓直徑低於500nm的黑色部分,則實施例25a在剩餘的黑色部分中,在同視場的二次電子像中存在碳材料的黑色部分沒有殘留,與此相對照,發電性能特別優良的實施例27a在剩餘的黑色部分中,如果測量在同視場的二次電子像中存在碳材料的黑色部分的個數,則為1個以上。因此,在實施例27a的催化劑層中,可以確認具有本發明特別優選的結構。此外,作為實施例27a具有上述結構的理由,可以列舉出實施例27a與實施例25a相比,凝聚相y中的碳材料b的含有率更高。
<性能評價結果5>
表8-1所示的實施例30a以及31a的催化劑層設定為如下的催化劑層結構:負極(鋅極)對置側的碳材料b的含有率α為0質量%以上且低於20質量%,多孔質擴散層側的碳材料b的含有率β超過10質量%且低於50質量%,且滿足α<β。實施例30a的催化劑層設定為2層結構,實施例31a設定為5層結構。
下面就實施例30a的催化劑層的製作方法進行說明。將疏水性多孔質層裁切成10cm見方(100cm2),首先,製作出pt濃度為0.25質量%的催化劑層油墨,以便成為在表8-1的實施例30a的下段所記載的組成,然後採用噴塗對該催化劑層油墨進行塗布,並在90℃下進行真空乾燥,對塗布量進行調整,從而使由塗布前後的疏水性多孔質層的質量變化算出的pt單位面積重量達到0.10mg/cm2。接著,製作出pt濃度為0.25質量%的催化劑層油墨,以便成為在表8-1的實施例30a的上段所記載的組成,然後採用噴塗對該催化劑層油墨進行塗布,並在90℃下進行真空乾燥,對塗布量進行調整,從而使由塗布前後的疏水性多孔質層的質量變化算出的pt單位面積重量達到0.10mg/cm2。其結果是,在疏水性多孔質層的mpl上,得到層疊有碳材料b的含有率為30質量%的催化劑層、和碳材料b的含有率為10質量%的催化劑層這2層的鉑單位面積重量為0.20mg/cm2的實施例30a的催化劑層。
在製作表8-1所示的實施例31a的催化劑層時,除了先前製作的40質量%的pt催化劑以外,還按照<催化劑的調配>中記載的方法,將碳材料c1作為催化劑載體的碳材料a,新調配擔載率為25質量%、30質量%、40質量%、50質量%的pt催化劑。使用擔載率為25質量%、30質量%、40質量%、50質量%的這4種pt催化劑,製作出5種催化劑層油墨,從而成為表8-1的實施例31a所示的5種(30質量%的催化劑使用2種)組成。對於從表8-1的實施例31a所記載的最下段的組成的催化劑層油墨起按順序的5種催化劑層油墨,將這些催化劑層油墨進行噴塗塗布,在90℃下進行真空乾燥,並使塗布前後的重量反覆發生變化,對於各自的油墨,一邊調整塗布量一邊進行再塗,從而使pt單位面積重量為0.04mg/cm2。其結果是,在疏水性多孔質層的mpl上,得到碳材料b的含有率從疏水性多孔質層側起從40質量%至0質量%階段性地發生變化的鉑單位面積重量為0.20mg/cm2的實施例31a的催化劑層。
關於這些實施例30a以及31a的催化劑層的性能試驗結果,得到了比表3~表7所示的所有比較例以及實施例更為優良的結果。其結果如表8-2所示。
表8-2
[實施例1b~36b以及比較例1b~30b]
<碳材料的物性測定>
在示出本發明的金屬空氣電池用電極的實施例1b~36b時,作為碳材料,準備12種碳材料a2~l2。表9(碳材料的種類和其物性)示出了各種碳材料的各種物性。
此外,表9所示的所述碳材料a2~l2各自的氮吸附比表面積sbet、總表面積stotal、微孔表面積smicro以及水蒸氣吸附量採用與對前述的碳材料a1~j1使用的測定方法同樣的方法進行了測定。此外,表9的dbp吸油量使用吸收儀(absorptiontometer,brabender公司生產),將最大轉矩的70%時的dbp添加量換算成每100g試料的dbp吸油量來決定。
<催化劑的調配>
將擔載著催化劑的碳材料作為碳材料a,從表9的碳材料中選擇1種作為碳材料a,除此以外,採用與前述的實施例1a~31a以及比較例1a~21a的催化劑的調配方法同樣的方法,調配擔載著pt的催化劑作為催化劑成分。
<催化劑層油墨的製作法1、2>
在製作本發明的金屬空氣電池用電極時,採用與前述的實施例1a~31a以及比較例1a~21a的塗布料漿的製作方法同樣的方法,製作出塗布料漿。也就是說,作為起始原料,使用按上述那樣製作的40質量%的催化劑以及從表9選擇的沒有擔載催化劑成分的碳材料,除此以外,採用與前述的實施例1a~31a以及比較例1a~21a的催化劑層油墨的製作方法即「催化劑層油墨的製作法1(或者2)同樣的方法,製作出催化劑層油墨。
此外,在催化劑層油墨的製作法2中,關於粉碎和攪拌處理的強度,可事先進行調整和決定,以便在使用按實施例1b的組成製作的催化劑層油墨而形成的催化劑層斷面的面積為10μm×10μm的視場中,使當量圓直徑在300nm以上這種大小的沒有催化劑成分的碳材料凝聚相(凝聚相y)分散1個以上,然後將其條件適用於所有的情況。
<電極形成>
除了使用按上述那樣製作的pt濃度為0.25質量%的催化劑層油墨以外,採用與前述的實施例1a~31a以及比較例1a~21a的空氣電池用正極的製作方法同樣的方法,製作出空氣電池用正極。另外,測量塗布前後的疏水性多孔質層的質量變化,算出pt單位面積重量,對塗布量進行調整,從而使鉑單位面積重量為0.20mg/cm2。
<金屬空氣電池性能評價用硬幣電池的製作>
為了評價得到的電極,使用按上述那樣製作的空氣電池用電極,除此以外,採用與前述的實施例1a~31a以及比較例1a~21a的金屬空氣電池性能評價用硬幣電池的製作方法同樣的方法,製作出實施例1b~36b以及比較例1b~30b的金屬空氣電池性能評價用硬幣電池。
<性能評價>
評價用硬幣電池在製作後,迅速地進行了性能評價。在評價中,將製作的評價用硬幣電池的下側殼體設定為負極,將上側帽蓋設定為正極,用能夠進行壓力控制的氣缸狀端子夾持硬幣電池,將氣缸端子的壓力控制為75kg/cm2,以便不會堵塞在帽蓋上形成的空氣孔,然後在室溫下以5ma的恆電流進行放電,將從放電開始10分鐘後的單電池電壓記錄為電池性能。
<性能評價結果1>
表10示出了使用由作為比較例的催化劑層油墨的製作法1調配的催化劑層油墨而形成的電極的組成、和編入了各自的電極的空氣電池的性能評價結果,表11示出了使用由催化劑層油墨的製作法2調配的催化劑層油墨而形成的電極的組成、和編入了各自的電極的空氣電池的性能評價結果。
此外,在表10和表11所示的所有情況下,擔載著催化劑的碳材料a使用滿足本發明的要件即水蒸氣吸附量和比smicro/stotal的碳材料b2,作為催化劑成分的pt的單位面積重量統一為0.20mg/cm2。另外,在使用未擔載催化劑的碳材料b的情況下,將種類統一為滿足作為本發明要件的水蒸氣吸附量和比x/sbet的碳材料k2,且催化劑層中的含量也統一為合計30質量%。另外,即使在含有粘結劑的情況下,種類統一為ptfe,催化劑層中的含有率統一為合計10wt%。
表11所示的實施例1b~4b無論在哪一種情況下,與表10以及表11所示的比較例1b~8b相比,顯示出更高的單電池電壓,顯示出更優良的電池特性。特別地,由沒有凝聚結構的方法製作的比較例1b~4b無論在哪一種情況下,單電池電壓不穩定,從放電開始電壓持續下降,10分鐘後低於0.8v。另外,對以具有凝聚結構的方式製作的比較例5b~8b、以及實施例1b~4b進行比較,凝聚相x中包含不具有與水的潤溼性的碳材料k2的實施例1b~4b與凝聚相x中不包含不具有與水的潤溼性的碳材料k2的比較例5b~8b相比較,顯示出較高的單電池電壓,具有優良的電池性能。如果在實施例1b~4b中進行比較,則完全不含有粘結劑的實施例1b在其中性能最差,僅在凝聚相y中含有粘結劑的實施例3b具有最優良的性能。
接著,就表10所示的比較例1b~4b的催化劑層、以及表11所示的實施例1b~4b的催化劑層進行了斷面結構觀察。對於觀察試料,用刀片以10mm左右見方的大小切出製作的電極,在用環氧進行樹脂填埋後,固定在冷凍切片機的夾具上,以便能夠切削催化劑層的斷面。將製作的夾具設置在切片機上,刀設置金剛石修整刀。此時,使金剛石修整刀相對於刀的行走方向偏移10度左右的角度,從而使催化劑層被傾斜地切削。
在修整後,繼續使用金剛石修整刀,沿催化劑層的深度方向以每1次50nm的速度至少切削100次,從而製作出催化劑層的切斷面。將製作了這些切斷面的催化劑層設置在電子顯微鏡上,以1萬倍的放大倍數對2次電子像和背散射電子圖像進行了觀察。在表10所示的比較例1b~4b的催化劑層中,背散射電子圖像以均勻明亮的對比度而被觀察到,沒有擔載催化劑成分的碳材料的凝聚相(氣體擴散碳材料凝聚相)沒有看到。與此相對照,表11所示的實施例1b~4b的催化劑層在由2次電子像可以明顯地判別存在碳材料的部位中,可以觀察到在背散射電子圖像中成為暗的對比度的部位、即沒有擔載催化劑成分的碳材料的凝聚相(氣體擴散碳材料凝聚相)。
為了更定量地進行識別,對於背散射電子圖像,在1萬倍的放大倍數下採用272dpi×272dpi以上的解析度、且以256色的層次的亮度採集作為電子信息,對於採集的圖像的亮度,使用圖像分析軟體進行二值化,從而將從暗的至第110層次的範圍用黑色表示,並使從第111層次向明的直至第256層次的範圍成為白色。接著,對各黑色點進行1次膨脹處理,從而對相鄰的點彼此之間進行識別。再者,執行填孔處理,對範圍內的空白部分進行填孔,從而識別為同一範圍。最後,進行使膨脹的部分復原的收縮處理,從而使目標的範圍明確化。而且由各黑色部分的面積算出各黑色部分的當量圓直徑,並將低於300nm的部分全部切除。在剩餘的黑色部分中,如果在同視場的二次電子像中測量存在碳材料的黑色部分的個數,則在實施例1b~4b的所有情況下為1個以上。
<性能評價結果2>
接著,在改變凝聚相x中含有的碳材料a的種類時進行了性能評價。表12示出了使用由催化劑層油墨的製作法2調配的催化劑層油墨而形成的電極的組成、和編入了各自的電極的空氣電池的性能評價結果。
此外,在表12所示的催化劑層中,作為催化劑成分的pt的單位面積重量統一為0.20mg/cm2,關於構成成分在催化劑層中的含有率,催化劑成分為22質量%,碳材料a為33質量%,碳材料b在凝聚相x中含有的部分為10質量%,在凝聚相y中含有的部分為15質量%,粘結劑不含在凝聚相x中,只含在凝聚相y中,其含有率統一為20質量%。凝聚相x中含有的碳材料b和凝聚相y中含有的碳材料b都使用滿足本發明要件的碳材料j2,粘結劑設定為ptfe。
碳材料b2、c2、d2、f2、g2、h2滿足與本發明的碳材料a有關的要件即25℃、相對壓力0.1的水蒸氣吸附量為0.1cm3/g以上、和採用氮吸附等溫線的t圖分析求出的微孔表面積smicro與總表面積stotal之比smicro/stotal為0.90以下這兩者,使用該碳材料b2、c2、d2、f2、g2、h2作為碳材料a的實施例5b~10b與使用並不滿足與本發明的碳材料a有關的要件的碳材料a2、e2、i2、j2、k2、l2的比較例9b~14b相比,可以產生更高的單電池電壓,從而顯示出優良的電池性能。特別地,比較例9b將微孔表面積處於支配地位且具有極高的水蒸氣吸附量的碳材料a2用作碳材料a,凝聚相x中含有與水的潤溼性較低的碳材料j2,並具有與專利文獻5的實施例9b類似的結構,該比較例9b在比較例中雖然具有較高的性能,但與實施例相比較,其性能較低。另外,即使在實施例中,實施例5b、6b、8b~10b將25℃、相對壓力0.1的水蒸氣吸附量為0.2cm3/g以上的碳材料b2、c2、f2、g2、h2用作碳材料a,該實施例5b、6b、8b~10b與使用25℃、相對壓力0.1的水蒸氣吸附量在0.1cm3/g以上且低於0.2cm3/g的碳材料d2作為碳材料a的實施例7b相比,具有更為優良的性能。
<性能評價結果3>
接著,在改變凝聚相x中含有的碳材料b的種類和催化劑層中的含有率時進行了性能評價。表13示出了使用由催化劑層油墨的製作法2調配的催化劑層油墨而形成的電極的組成、和編入了各自的電極的空氣電池的性能評價結果。
此外,在表13所示的催化劑層中,作為催化劑成分的pt的單位面積重量統一為0.20mg/cm2,關於碳材料b在催化劑層中的含有率,凝聚相y中含有的部分為10質量%,粘結劑不含在凝聚相x中,只含在凝聚相y中,統一含有10質量%。碳材料a使用滿足本發明要件的碳材料g2,凝聚相y中含有的碳材料b使用滿足本發明要件的碳材料i2,粘結劑使用ptfe。
碳材料e2、i2、j2、k2、l2滿足與本發明的碳材料b有關的要件即25℃、相對壓力0.1下的水蒸氣吸附量低於0.1cm3/g,dbp吸油量x(cm3/100g)和基於bet評價的比表面積sbet之比x/sbet為0.5以上這兩者,將該碳材料e2、i2、j2、k2、l2用作在凝聚相x中含有的碳材料b的實施例11b~21b和將並不滿足與本發明的碳材料b有關的要件的碳材料a2、b2、c2、d2、f2、g2、h2用作凝聚相x中含有的碳材料b的比較例15b~22b相比,可以產生更高的單電池電壓,從而顯示出優良的電池性能。特別地,在將水蒸氣吸附量低於0.05cm3/l的碳材料k2、l2用作碳材料b的實施例20b、21b中,電池特性特別良好。另外,在表13中,對使凝聚相x中含有的碳材料b在催化劑層中的含有率於0~40質量%之間發生變化的比較例22b、以及實施例12b~18b進行了比較。其結果是,凝聚相x中不含有碳材料b的比較例22b、凝聚相x中含有40質量%的碳材料b、且凝聚相y中也含有10質量%的碳材料b、從而催化劑層中的碳材料b的合計含有率為50質量%的實施例18b與凝聚相x中和凝聚相y中都含有碳材料b、催化劑層中的合計含有率為5質量%以上且低於50質量%的實施例12b~17b相比,為較差的性能。另外,在催化劑層中的合計含有率為10質量%~40質量%的情況下,其特性變得特別良好。另外,在凝聚相x中的碳材料b的含有率為0.1~0.5的實施例13b~17b中,其特性變得特別良好。
<性能評價結果4>
接著,在改變凝聚相y中含有的碳材料b的種類和催化劑層中的含有率時進行了性能評價。表14示出了使用由催化劑層油墨的製作法2調配的催化劑層油墨而形成的電極的組成、和編入了各自的電極的空氣電池的性能評價結果。
此外,在表14所示的催化劑層中,作為催化劑成分的pt的單位面積重量統一為0.20mg/cm2;關於碳材料b在催化劑層中的含有率,凝聚相x中含有的部分為5質量%;粘結劑不含在凝聚相x中,只含在凝聚相y中,催化劑層中的含有率統一為10質量%。特別地,在實施例25b~27b中,將凝聚相y中的碳材料b在催化劑層中的含有率固定為3質量%,使凝聚相x中的碳材料b在催化劑層中的含有率發生變化。碳材料a使用滿足本發明要件的碳材料c2,凝聚相x中含有的碳材料b使用滿足本發明要件的碳材料k2,粘結劑使用ptfe。
碳材料e2、i2、j2、k2、l2滿足與本發明的碳材料b有關的要件即25℃、相對壓力0.1下的水蒸氣吸附量低於0.1cm3/g,dbp吸油量x(cm3/100g)和基於bet評價的比表面積sbet之比x/sbet為0.5以上這兩者,將該碳材料e2、i2、j2、k2、l2用作在凝聚相y中含有的碳材料b的實施例22b~34b和將並不滿足與本發明的碳材料b有關的要件的碳材料a2、b2、c2、d2、f2、g2、h2用作凝聚相y中含有的碳材料b的比較例23b~29b相比,可以產生更高的單電池電壓,從而顯示出優良的電池性能。另外,在表14中,如果對使凝聚相y中含有的碳材料b在催化劑層中的含有率於0~45質量%之間發生變化的比較例30b、以及實施例25b~33b進行比較,則凝聚相y中不含有碳材料b的比較例30b,在凝聚相x中含有1質量%的碳材料b、在凝聚相y中含有3質量%的碳材料b、催化劑層中的碳材料b的合計含有率低於5質量%的實施例25b,在凝聚相x中含有5質量%的碳材料b、在凝聚相y中也含有45質量%的碳材料b、催化劑層中的碳材料b的合計含有率為50質量%以上的實施例33b與在凝聚相x和凝聚相y中均含有碳材料b、催化劑層中的合計含有率為5質量%以上且低於50質量%的實施例26b~32b相比,為較差的性能。
另外,在表14所示的催化劑層中,對於實施例28b和30b的催化劑層,採用與<性能評價結果1>同樣的方法,進行了斷面結構觀察,結果在300nm以上的黑色部分中,如果在同視場的二次電子像中測量存在碳材料的黑色部分的個數,則確認為1個以上。再者,如果削除當量圓直徑低於500nm的黑色部分,則實施例28b在剩餘的黑色部分中,在同視場的二次電子像中存在碳材料的黑色部分沒有殘留,與此相對照,發電性能特別優良的實施例30b在剩餘的黑色部分中,如果測量在同視場的二次電子像中存在碳材料的黑色部分的個數,則為1個以上。因此,在實施例30b的催化劑層中,可以確認具有本發明特別優選的結構。此外,作為實施例30b具有上述結構的理由,可以列舉出實施例30b與實施例28b相比,凝聚相y中的碳材料b的含有率更高。
<性能評價結果5>
表15-1所示的實施例35b以及36b的催化劑層設定為如下的催化劑層結構:負極(鋅極)對置側的碳材料b的含有率α為0質量%以上且低於20質量%,多孔質擴散層側的碳材料b的含有率β超過10質量%且低於50質量%,且滿足α<β。實施例35b的催化劑層設定為2層結構,實施例36b設定為5層結構。此外,實施例35b以及36b的凝聚相x使用滿足本發明要件的碳材料c2作為碳材料a,使用滿足本發明要件的碳材料k2作為碳材料b。另外,實施例35b以及36b的凝聚相y使用所述碳材料k2作為碳材料b。
下面就實施例35b的催化劑層的製作方法進行說明。將疏水性多孔質層裁切成10cm見方(100cm2),首先,製作出pt濃度為0.25質量%的催化劑層油墨,以便成為在表15-1的實施例35b的下段所記載的組成,然後採用噴塗對該催化劑層油墨進行塗布,並在90℃下進行真空乾燥,對塗布量進行調整,從而使由塗布前後的疏水性多孔質層的質量變化算出的pt單位面積重量達到0.10mg/cm2。接著,製作出pt濃度為0.25質量%的催化劑層油墨,以便成為在表15-1的實施例35b的上段所記載的組成,然後採用噴塗對該催化劑層油墨進行塗布,並在90℃下進行真空乾燥,對塗布量進行調整,從而使由塗布前後的疏水性多孔質層的質量變化算出的pt單位面積重量達到0.10mg/cm2。其結果是,在疏水性多孔質層的mpl上,得到層疊有碳材料b的含有率為30質量%的催化劑層、和碳材料b的含有率為10質量%的催化劑層這2層的鉑單位面積重量為0.20mg/cm2的實施例35b的催化劑層。
在製作表15-1所示的實施例36b的催化劑層時,除了先前製作的40質量%的pt催化劑以外,還按照<催化劑的調配>中記載的方法,將碳材料c2作為催化劑載體的碳材料a,新調配擔載率為25質量%、30質量%、40質量%、50質量%的pt催化劑。使用擔載率為25質量%、30質量%、40質量%、50質量%的這4種pt催化劑,製作出5種催化劑層油墨,從而成為表15-1的實施例36b所示的5種(30質量%的催化劑使用2種)組成。對於從表15-1的實施例36b所記載的最下段的組成的催化劑層油墨起按順序的5種催化劑層油墨,將這些催化劑層油墨進行噴塗塗布,在90℃下進行真空乾燥,並使塗布前後的重量反覆發生變化,對於各自的油墨,一邊調整塗布量一邊進行再塗,從而使pt單位面積重量為0.04mg/cm2。其結果是,在疏水性多孔質層的mpl上,得到碳材料b的含有率從疏水性多孔質層側起從40質量%至0質量%階段性地發生變化的鉑單位面積重量為0.20mg/cm2的實施例36b的催化劑層。
關於這些實施例35b以及36b的催化劑層的性能試驗結果,得到了比表10~表14所示的所有比較例以及實施例更為優良的結果。其結果如表15-2所示。
表15-2
以上參照附圖,就本發明優選的實施方式進行了詳細的說明,但本發明並不限定於這樣的例子。只要是具有本發明所屬技術領域的通常的知識的人員,在權利要求書所記載的技術思想的範疇內,顯然可以想到各種變更例或修正例,對於這些,當然可以理解為也屬於本發明的技術範圍。
符號說明:
1負極
2隔膜或者電解質膜
3正極
4催化劑層
5多孔質層
6電解質水溶液
7集電體(負極)
8集電體(正極)
9電解質水溶液和空氣的界面(可期待三相界面的部位)
10ptfe或者蠟
11疏水性碳材料(在本發明中為碳材料b)
12擔載著催化劑成分的碳材料
13電解質材料
14導電助劑碳材料
15氣體擴散碳材料
16催化劑凝聚相
17氣體擴散凝聚相
18擔載著催化劑成分的碳材料a
19碳材料b
20凝聚相x
21凝聚相y
22碳材料a
23催化劑成分