一種檢測水中極性苯酚類氯代/溴代消毒副產物的方法與流程
2023-06-05 18:32:36 2
本發明涉及極性苯酚類滷代消毒副產物的檢測和定量,尤其涉及一種檢測水中痕量極性苯酚類氯代/溴代消毒副產物的檢測和定量方法。
背景技術:
自從上世紀80年代人們首次發現飲用水消毒副產物以來,文獻中所報導的飲用水消毒副產物已有約600-700種[1]。然而,由於檢測手段的局限性,過去的大多數研究都集中在一些常規的脂肪族消毒副產物(如三滷甲烷、滷代乙酸等)上,對極性苯酚類氯代/溴代消毒副產物的研究相對較少。
現有研究基於超效液相色譜/電噴霧三重四極杆質譜技術的前體離子掃描的方法、多反應監測和子離子掃描功能的結合[4][5],在飲用水中發現了13種新型極性氯代/溴代苯酚類消毒副產物,例如:3,5-二氯-4-羥基苯甲醛、3-溴-5-氯-4-羥基苯甲醛、3,5-二溴-4-羥基苯甲醛、3,5-二氯-4-羥基苯甲酸、3-溴-5-氯-4-羥基苯甲酸、3,5-二溴-4-羥基苯甲酸、3,5-二氯水楊酸、3-溴-5-氯水楊酸、3,5-二溴水楊酸、2,4,6-三氯苯酚、2,6-二氯-4-溴苯酚、2,6-二溴-4-氯苯酚和2,4,6-三溴苯酚等[6][7]。然而,這種檢測方法耗時長,每次只能檢測一種苯酚類消毒副產物,且該方法檢出限(2.87-136.78ng/L)和定量限(6.24-199.20ng/L)比較高,很大程度地限制了針對僅含痕量(一般10-12-10-6g/L範圍內)的新型極性氯代/溴代苯酚類消毒副產物的水體的研究。然而,相關毒理學研究顯示[2][3],新型極性苯酚類氯代/溴代消毒副產物比常規脂肪族消毒副產物具有更高的生長發育毒性和生長抑制作用。因此,開發一種能夠快速高效、高靈敏度地檢測水體中13種新型極性苯酚類氯代/溴代消毒副產物的方法顯得尤為必要。
參考文獻
[1]Zhang,X.R.;Minear,R.A.;Guo,Y.B.;Hwang,C.J.;Barrett,S.E.;Ikeda,K.;Shimizu,Y.;Matsui,S.,Occurrence,genotoxicity,and carcinogenicity of regulated and emerging disinfection by-products in drinking water:A review and roadmap for research.Mutat Res-Rev Mutat,2007,636,(1-3):178-242.
[2]Yang,M.T.;Zhang,X.R.,Comparative Developmental Toxicity of New Aromatic Halogenated DBPs in a Chlorinated Saline Sewage Effluent to the Marine Polychaete Platynereis dumerilii.Environ Sci Technol,2013,47,(19):10868-10876.
[3]Liu,J.Q.;Zhang,X.R.,Comparative toxicity of new halophenolic DBPs in chlorinated saline wastewater effluents against a marine alga:Halophenolic DBPs are generally more toxic than haloaliphatic ones.Water Research,2014,65:64-72.
[4]Zhang,X.R.;Talley,J.W.;Boggess,B.;Ding,G.Y.;Birdsell,D.,An electrospray ionization-tandem mass spectrometry method for identifying chlorinated drinking water disinfection byproducts.Water Research,2004,38,(18):3920-3930.
[5]Zhang,X.R,et al.Fast selective detection of polar brominated disinfection byproducts in drinking water using precursor ion scans.Environ Sci Technol,2008,42,(17):6598-6603.
[6]Zhai,H.Y.;Zhang,X.R.,Formation and Decomposition of New and Unknown Polar Brominated Disinfection Byproducts during Chlorination.Environ Sci Technol,2011,45,(6):2194-2201.
[7]Pan,Y.;Zhang,X.R.,Four Groups of New Aromatic Halogenated Disinfection Byproducts:Effect of Bromide Concentration on Their Formation and Speciation in Chlorinated Drinking Water.Environ Sci Technol,2013,47,(3):1265-1273.
技術實現要素:
本發明的目的在於提供一種檢測水中13種新型極性苯酚類氯代/溴代消毒副產物的方法,以解決現有檢測方法耗時長、檢出限和定量限比較高,無法應用於檢測水體中所含有的痕量極性苯酚類氯代/溴代消毒副產物的技術問題。
為了實現上述目的,本發明的技術方案如下:
本發明提供了一種檢測極性苯酚類氯代/溴代消毒副產物的方法,包括以下步驟:
1)配製標準工作溶液:配製不同濃度梯度的含3,5-二氯-4-羥基苯甲醛、3-溴-5-氯-4-羥基苯甲醛、3,5-二溴-4-羥基苯甲醛、3,5-二氯-4-羥基苯甲酸、3-溴-5-氯-4-羥基苯甲酸、3,5-二溴-4-羥基苯甲酸、3,5-二氯水楊酸、3-溴-5-氯水楊酸、3,5-二溴水楊酸、2,4,6-三氯苯酚、2,6-二氯-4-溴苯酚、2,6-二溴-4-氯苯酚、2,4,6-三溴苯酚的標準工作溶液,該標準工作溶液用於標準加入法來測定樣品中待測物質的濃度;可選地,所述的濃度梯度為10-12g/L-10-6g/L;
2)待測水樣的預處理:對待測水樣進行連續液液萃取、酸化、鹽析、分離、濃縮和過濾等預處理;
可選地,對單位體積的待測水樣用不參與反應的無機酸(如:H2SO4、HCl或者HNO3等)將pH調至≤0.5,隨後在此酸化過的待測水樣中加入易溶於水但不參與反應的無機鹽(如:亞砷酸鈉、硼氫化鈉、硫酸銨、亞硫酸鈉、氯化銨、抗壞血酸或者硫代硫酸鈉等)使其飽和,然後再加入0.1單位體積的有機萃取劑,即:甲基叔丁基醚、乙酸乙酯、正戊烷、環己烷或者正己烷中的一種,進行萃取,放置至少10min後放出水相,再用0.05單位體積的上述萃取步驟中所用的有機萃取劑對上層有機相進行第二次萃取,靜止至少5min後,再將上層有機相濃縮至0.5mL,可選地,採用旋轉蒸發儀減壓濃縮或者使用氮氣吹脫濃縮,隨後向該濃縮有機相中加入0.01單位體積的乙腈,隨後再次濃縮至0.5mL,可選地,採用旋轉蒸發儀減壓濃縮或者使用氮氣吹脫濃縮;最後,將其用超純水稀釋至1mL,並用孔徑≤0.45μm的聚四氟乙烯膜過濾;
優選地,對待測水樣用H2SO4將pH調到0.5,隨後對於每升酸化過的樣品加入100g Na2SO4使其飽和,然後再加入100mL甲基叔丁基醚萃取,放置10-30min後用50mL甲基叔丁基醚對上層有機層進行二次萃取,靜止5-10min後再將上層有機層轉移到旋轉蒸發儀中並且減壓濃縮到0.5mL,隨後加入10mL的乙腈旋轉蒸發到0.5mL;最後將上述經過酸化、加Na2SO4和多次萃取旋蒸後的待測水樣用超純水稀釋至1mL,並用孔徑為0.45μm的聚四氟乙烯膜過濾;
3)超高效液相色譜/電噴霧電離三重四極杆質譜儀(UPLC/ESI-tqMS)分析:建立UPLC/ESI-tqMS的質譜參數、液相色譜參數和梯度洗脫過程參數,檢測13種苯酚類氯代/溴代消毒副產物中的任意一種或者幾種;其中:
質譜參數為:電離方式,電噴霧電離負離子模式;毛細管電壓,2.5-3.7kV;源溫度,100-150℃;脫溶劑溫度,300-650℃;脫溶劑氣流速,600-1000L/h;錐孔氣流速,25-200L/h;
液相色譜參數:進樣體積,2-10μL;色譜柱型號,Acquity UPLC HSS T3、HSS C18、HSS PFP(2.1×100mm,填充物粒徑1.8μm,Waters);柱溫,20-40℃;流動相,水/甲醇和水/乙腈;流速,0.4mL/min;
梯度洗脫過程如下:在開始的8min裡,流動相水/甲醇的比例從95/5線性的變成5/95,在之後的0.1min裡面線性的變回95/5,這個組分繼續保持2.9min,使柱子平衡;
4)繪製校正曲線和結果計算:在步驟3)中得到的色譜圖的基礎上,初步判定樣品中的色譜峰是否與已知標準物質為同一物質,可選地,所採用的判定方法為已知物質保留值直接對照法、增加峰高法或三維圖譜檢測器定性法;在確定為同一種物質後,再分別計算(如:採用標準加入法)待測水樣中的13種新型極性苯酚類氯代/溴代消毒副產物的濃度。
其中標準加入法具體步驟為:配製濃度為CX+0、CX+C1、CX+C2、CX+C3和CX+C4的和樣品有相同基體的標準系列溶液(其中CX代表待測樣品中待測物質的濃度,C1、C2、C3和C4代表一定濃度的待測物質的標準溶液),並對這些樣品重複做UPLC/ESI-tqMS多反應監測掃描分析得到峰面積為S0、S1、S2、S3和S3的色譜圖。以CX+0、CX+C1、CX+C2、CX+C3和CX+C4橫坐標,以S0、S1、S2、S3和S3為縱坐標繪製標準加入法的校正曲線,求得校正曲線的方程式為y=kx+CX;將校正曲線外延與橫坐標相交,原點至交點的距離,即為試樣中待測元素的含量CX。
本發明的技術方案達到了如下的有益效果:
1)高靈敏度:本發明採用連續液液萃取手段能有效地萃取出樣品中的待測物,並建立檢測方法相關的儀器參數,從而大大的提高了檢測方法的靈敏度。以1L水樣計,本發明對13種新型極性苯酚類氯代/溴代消毒副產物的檢出限的最優範圍為0.42-26.64ng/L,遠低於常規方法(具體方法見背景技術中的參考文獻[3])的2.87-136.78ng/L;定量限範圍為1.35-63.64ng/L,遠低於常規方法的6.24-199.20ng/L。具體數值見表2。
2)高精密度和準確度:本方法採用標準加入法進行定量,減少了操作過程中因樣品溶液基底不同帶來的誤差。大大的提升了前處理方法的重現性和回收率,並且降低了儀器精密度帶來的誤差。以1L水樣計,本發明對濃度為1mg/L的13種新型極性苯酚類氯代/溴代消毒副產物的混合溶液測量相對標準偏差範圍為2.2%-3.9%,遠低於常規方法的10%;回收率範圍為95%-103%,遠低於常規方法的90%-110%。
3)快速:同時測定13個樣品所需最少總時間僅為11min,遠低於常規方法(具體方法見背景技術中的參考文獻[3])所需總時間143min。
本發明的檢測方法具有高靈敏度、高密精密度和準確度,可實現快速高效地測定水體中極性苯酚類氯代/溴代消毒副產物的目的,尤其是水中痕量極性苯酚類氯代/溴代消毒副產物的檢測,具有較高的實際應用價值。
附圖說明
圖1是13種新型極性氯代/溴代消毒副產物的超高效液相色譜電噴霧電離三重四極杆質譜多反應監測掃描色譜圖。
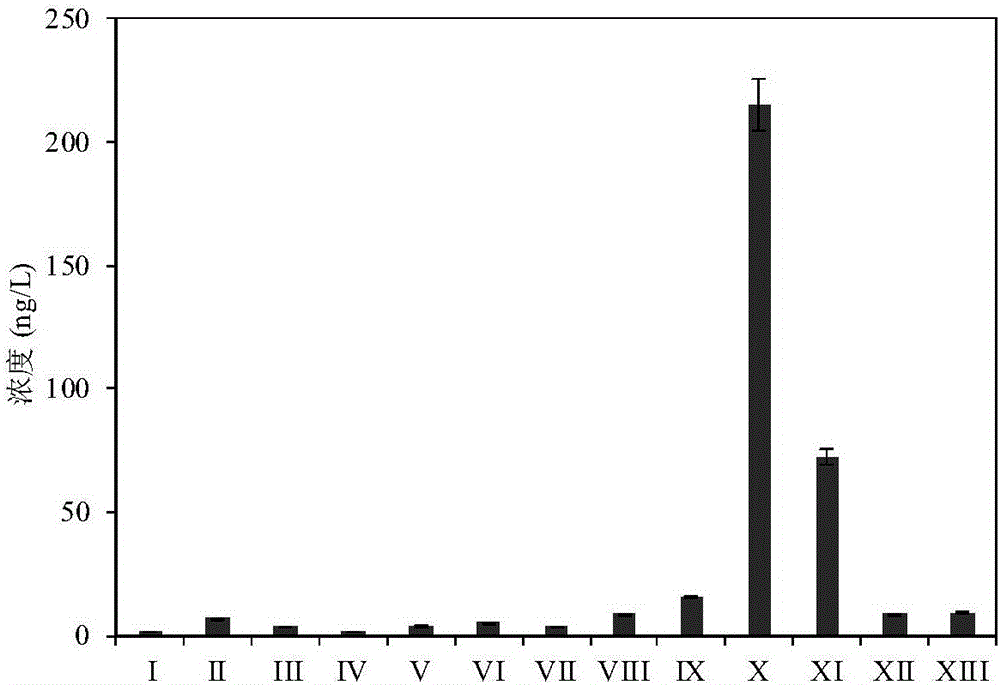
圖2是13種新型極性苯酚類氯代/溴代消毒副產物在自來水廠出廠水中的濃度分布圖。
圖3是13種新型極性苯酚類氯代/溴代消毒副產物在城市二級汙水處理廠出廠水消毒實驗中的濃度分布圖。
圖4是13種新型極性苯酚類氯代溴代消毒副產物在模擬飲用水樣消毒實驗中的濃度分布圖。
圖中,I.3,5-二氯-4-羥基苯甲醛,II.3-溴-5-氯-4-羥基苯甲醛,III.3,5-二溴-4-羥基苯甲醛,IV.3,5-二氯-4-羥基苯甲酸,V.3-溴-5-氯-4-羥基苯甲酸,VI.3,5-二溴-4-羥基苯甲酸,VII.3,5-二氯水楊酸,VIII.3-溴-5-氯水楊酸,IX.3,5-二溴水楊酸,X.2,4,6-三氯苯酚,XI.2,6-二氯-4-溴苯酚,XII.2,6-二溴-4-氯苯酚,XIII.2,4,6-三溴苯酚。
具體實施方式
為了闡明本發明的技術方案及技術目的,下面結合附圖及具體實施方式對本發明做進一步的介紹。
實施例1
圖1顯示了本發明的技術方案可以一次同時實現13種新型極性苯酚類氯代/溴代消毒副產物的分離與檢測。其具體方法包括如下步驟:
1)配製不同濃度梯度(10-12g/L-10-6g/L)的含3,5-二氯-4-羥基苯甲醛、3-溴-5-氯-4-羥基苯甲醛、3,5-二溴-4-羥基苯甲醛、3,5-二氯-4-羥基苯甲酸、3-溴-5-氯-4-羥基苯甲酸(以體積計)、3,5-二溴-4-羥基苯甲酸、3,5-二氯水楊酸、3-溴-5-氯水楊酸、3,5-二溴水楊酸、2,4,6-三氯苯酚、2,6-二氯-4-溴苯酚、2,6-二溴-4-氯苯酚、2,4,6-三溴苯酚的標工作準溶液。其中,3-溴-5-氯-4-羥基苯甲酸沒有可用的標準品,因而根據文獻(Pan,Y.,Zhang,X.,2013.Four groups of new aromatic halogenated disinfection byproducts:Effect of bromide concentration on their formation and speciation in chlorinated drinking water.Environmental Science and Technology 47(3),1265–1273)中的方法自己合成,其餘的均可在網上買到標準品。
2)配製含3,5-二氯-4-羥基苯甲醛、3-溴-5-氯-4-羥基苯甲醛、3,5-二溴-4-羥基苯甲醛、3,5-二氯-4-羥基苯甲酸、3,5-二溴-4-羥基苯甲酸、3,5-二氯水楊酸、3-溴-5-氯水楊酸、3,5-二溴水楊酸、2,4,6-三氯苯酚、2,6-二氯-4-溴苯酚、2,6-二溴-4-氯苯酚、2,4,6-三溴苯酚的濃度為1mg/L混合溶液作為待測水樣。
3)取步驟2)中所配置的待測水樣1L用H2SO4將pH調到0.5。對於每升酸化過的樣品加入100g Na2SO4使其飽和,然後再加入100mL甲基叔丁基醚萃取,放置15min後用50mL甲基叔丁基醚對上層有機層進行二次萃取,靜止5min後再將上層有機層轉移到旋轉蒸發儀中並且減壓濃縮到0.5mL,在加入10mL的乙腈旋轉蒸發到0.5mL。將上述經過酸化、加Na2SO4、多重萃取和旋蒸後的待測水樣用超純水稀釋至1mL,並用孔徑為0.45μm的聚四氟乙烯膜過濾。
4)對步驟1)中所提及的13種苯酚類氯代/溴代消毒副產物進行一次同時檢測:建立的超高效液相色譜/電噴霧電離三重四極杆質譜儀(UPLC/ESI-tqMS)檢測分析方法的參數設置如下:a.質譜參數:電離方式,電噴霧電離負離子模式;毛細管電壓,3.1kV;源溫度,130℃;脫溶劑溫度,500℃;脫溶劑氣流速,800L/h;錐孔氣流速,50L/h;13種化合物的多反應監測掃描方法所涉及的碰撞能、錐孔電壓和離子駐留時間見表1;b.液相色譜參數:進樣體積,5μL;色譜柱型號,Acquity UPLC HSS T3(2.1×100mm,填充物粒徑1.8μm,Waters);柱溫,30℃;流動相,水/甲醇;流速,0.4mL/min。梯度洗脫過程如下:在開始的8min裡,流動相水/甲醇的比例從95/5線性的變成5/95,在之後的0.1min裡面線性的變回95/5,這個組分繼續保持2.9min,使柱子平衡。
表1:13種新型極性苯酚類氯代/溴代消毒副產物的錐孔電壓、碰撞能和離子駐留時間。
5)對步驟4)中得到的色譜圖採用已知物質保留值直接對照法、增加峰高法或三維圖譜檢測器定性法,初步判定樣品中的色譜峰是否與已知標準物質為同一物質。在確定是同一種物質後,採用標準加入法分別計算樣品中的13種新型極性苯酚類氯代/溴代消毒副產物的濃度。
本實施例1中測定這13種新型極性苯酚類氯代/溴代消毒副產物所需總時間為11min。
隨後,根據13種新型極性苯酚類氯代/溴代消毒副產物濃度檢測結果,對本方法的檢出限、定量限、精密度和準確度進行了確定,具體方法如下:
1)檢出限和定量限的確定方法:每種新型極性苯酚類氯代/溴代消毒副產物的測定值分別能滿足7次測定值相對標準偏差≤10%、回收率在90%至110%之間、信噪比≥3(檢出限)和信噪比≥10(定量限)的最低濃度。(用於測定的溶液體積為1L,由13種新型極性苯酚類氯代/溴代消毒副產物標準品配製。)
2)精密度的確定方法:用被測物質標準品定量稱樣並配製成相對濃度為80%、100%、120%三種測試溶液,每種溶液製備3份,每份溶液進樣三次。計算每個樣品三次進樣的平均峰面積及RSD;計算每個濃度點的3份溶液的含量及相應的RSD;計算每個濃度點的平均含量及3個點間相應的RSD。
3)準確度的確定方法:用被測物質標準品定量稱樣並配製成相對濃度為80%、100%、120%三種測試溶液,每種溶液製備3份,每份溶液進樣三次。計算每個樣品溶液的平均含量,並計算3個濃度點的9個溶液的總平均含量值與大於95%的置信區間。
本實施例1中,檢出限和定量限範圍分別為0.42-26.64ng/L和1.35-63.64ng/L(詳見表2)。相對標準偏差範圍為1.9%-2.2%;回收率範圍為98%-103%。
表2:13種新型極性苯酚類氯代/溴代消毒副產物的檢測方法的檢出限和定量限、相對標準偏差和回收率。
實施例2
在實施例2中,針對13種新型極性苯酚類氯代/溴代消毒副產物中的一種或多種物質採取本發明的技術方案同時進行測定。其具體方法包括如下步驟:
步驟1)配製不同濃度梯度的標準工作溶液,2)配置待測水樣,以及步驟3)待測水樣的預處理,與實施例1中所描述的方法相同。
步驟4)中,對13種苯酚類氯代/溴代消毒副產物中的進行了隨機組合,即對13種苯酚類氯代/溴代消毒副產物中的一種或者幾種進行組合,分次(2-12次)採用本發明中的檢測方法進行檢測。建立的超高效液相色譜/電噴霧電離三重四極杆質譜儀(UPLC/ESI-tqMS)檢測分析方法的參數設置與實施例1中的相關參數設置一樣。
步驟5)中,對步驟4)中得到的色譜圖採用已知物質保留值直接對照法、增加峰高法或三維圖譜檢測器定性法,初步判定樣品中的色譜峰是否與已知標準物質為同一物質。確定是同一種物質後再採用標準加入法分別計算樣品中的13種新型極性苯酚類氯代/溴代消毒副產物的濃度。
根據本實施例中選取同時檢測的樣品的數量的不同,本實施例中測定這13種新型極性苯酚類氯代/溴代消毒副產物所需總時間為22-143min。
本實施例中此方法的檢出限和定量限範圍分別為0.42-26.64ng/L和1.35-63.64ng/L(詳見表2)。
實施例3
在實施例3中,採用本發明的檢測方法對自來水廠出廠水中所含的13種新型極性苯酚類氯代/溴代消毒副產物進行了分離與檢測。其具體方法包括如下步驟:
1)配製不同濃度梯度的標準工作溶液與實施例1中所描述的方法相同。
2)採集新鮮的自來水廠出廠水1L,水樣在4℃冷藏條件下快速運回實驗室。
3)取2)中水樣100mL滴定水中餘氯含量,然後向剩餘的0.9L水樣中加入摩爾比過量5%的亞砷酸鈉終止反應。再用H2SO4將pH調到0.5。對於每升酸化過的樣品加入90g Na2SO4使其飽和,然後再加入90mL甲基叔丁基醚萃取,放置15min後用45mL甲基叔丁基醚對上層有機層進行二次萃取,靜止5min後再將上層有機層轉移到旋轉蒸發儀中並且減壓濃縮到0.5mL,在加入10mL的乙腈旋轉蒸發到0.5mL。將上述經過酸化、加Na2SO4、多重萃取和旋蒸後的待測水樣用超純水稀釋至1mL,並用孔徑為0.45μm的聚四氟乙烯膜過濾。
4)採用本發明中的檢測方法同時對13種苯酚類氯代/溴代消毒副產物進行檢測。建立的超高效液相色譜/電噴霧電離三重四極杆質譜儀(UPLC/ESI-tqMS)檢測分析方法的參數設置如下:a.質譜參數:電離方式,電噴霧電離負離子模式;毛細管電壓,3.1kV;源溫度,130℃;脫溶劑溫度,500℃;脫溶劑氣流速,800L/h;錐孔氣流速,50L/h;13種化合物的多反應監測掃描方法所涉及的碰撞能、錐孔電壓和離子駐留時間見表1。b.液相色譜參數:進樣體積,5μL;色譜柱型號,Acquity UPLC HSS T3(2.1×100mm,填充物粒徑1.8μm,Waters);柱溫,30℃;流動相,水/甲醇;流速,0.4mL/min。梯度洗脫過程如下:在開始的8min裡,流動相水/甲醇的比例從95/5線性的變成5/95,在之後的0.1min裡面線性的變回95/5,這個組分繼續保持2.9min,使柱子平衡。
5)對步驟4)中得到的色譜圖採用已知物質保留值直接對照法、增加峰高法或三維圖譜檢測器定性法,初步判定樣品中的色譜峰是否與已知標準物質為同一物質。確定是同一種物質後再採用標準加入法分別計算樣品中的13種新型極性苯酚類氯代/溴代消毒副產物的濃度。
本實施例中測定這13種新型極性苯酚類氯代/溴代消毒副產物所需總時間為11min。
本實施例中檢測出13種待測消毒副產物(見圖2),其中3,5-二氯-4-羥基苯甲醛濃度低於方法定量限。其餘12種待測消毒副產物的濃度範圍為1.35–215.0ng/L。
實施例4
在實施例4中,採用本發明的檢測方法對城市二級汙水處理廠出廠水中所含的13種新型極性苯酚類氯代/溴代消毒副產物進行了分離與檢測。其具體方法包括如下步驟:
1)配製不同濃度梯度的標準工作溶液與實施例1中所描述的方法相同。
2)採集新鮮的城市二級汙水處理廠出廠水1L,水樣在4℃冷藏條件下快速運回實驗室,在實驗室分別用5mg的氯胺或氯(以次氯酸鈉代替)(以Cl2計)消毒12h。
3)取2)中經消毒12h的水樣100mL滴定水中餘氯含量,然後向剩餘的0.9L水樣中加入摩爾比過量5%的氯化銨溶液終止反應。再用H2SO4將pH調到0.5。對於每升酸化過的樣品加入90g Na2SO4使其飽和,然後再加入90mL甲基叔丁基醚萃取,放置15min後用45mL甲基叔丁基醚對上層有機層進行二次萃取,靜止5min後再將上層有機層轉移到旋轉蒸發儀中並且減壓濃縮到0.5mL,在加入10mL的乙腈旋轉蒸發到0.5mL。將上述經過酸化、加Na2SO4、多重萃取和旋蒸後的待測水樣用超純水稀釋至1mL,並用孔徑為0.45μm的聚四氟乙烯膜過濾。
4)採用本發明中的檢測方法同時對13種苯酚類氯代/溴代消毒副產物進行檢測。建立的超高效液相色譜/電噴霧電離三重四極杆質譜儀(UPLC/ESI-tqMS)檢測分析方法的參數設置如下:a.質譜參數:電離方式,電噴霧電離負離子模式;毛細管電壓,3.1kV;源溫度,130℃;脫溶劑溫度,500℃;脫溶劑氣流速,800L/h;錐孔氣流速,50L/h;13種化合物的多反應監測掃描方法所涉及的碰撞能、錐孔電壓和離子駐留時間見表1;b.液相色譜參數:進樣體積,5μL;色譜柱型號,Acquity UPLC HSS T3(2.1×100mm,填充物粒徑1.8μm,Waters);柱溫,30℃;流動相,水/甲醇;流速,0.4mL/min。梯度洗脫過程如下:在開始的8min裡,流動相水/甲醇的比例從95/5線性的變成5/95,在之後的0.1min裡面線性的變回95/5,這個組分繼續保持2.9min,使柱子平衡。
5)對步驟4)中得到的色譜圖採用已知物質保留值直接對照法、增加峰高法或三維圖譜檢測器定性法,初步判定樣品中的色譜峰是否與已知標準物質為同一物質。確定是同一種物質後再採用標準加入法分別計算樣品中的13種新型極性苯酚類氯代/溴代消毒副產物的濃度。
本實施例4中測定這13種新型極性苯酚類氯代/溴代消毒副產物所需總時間為11min。
在本實施例4針對氯胺消毒和氯消毒的城市二級汙水處理廠出廠水中,均檢測到此13種新型極性苯酚類氯代/溴代消毒副產物(見圖3),所檢測到的濃度範圍為分別為8.98-980.4ng/L和15.36-1900.5ng/L。
實施例5
在實施例5中,採用本發明的檢測方法對實驗室配製1L模擬飲用水樣中所含的13種新型極性苯酚類氯代/溴代消毒副產物進行了分離與檢測。其具體方法包括如下步驟:
1)配製不同濃度梯度的標準工作溶液與實施例1中所描述的方法相同。
2)在實驗室配製1L模擬飲用水樣:將3mg(以C計)的天然有機物(SRNOM,2SRFA或SRHA或2R101N),90mg(以CaCO3計)的NaHCO3和400μg(以Br計)的KBr溶解於1L超純水中。分別用5mg的氯胺或氯(以次氯酸鈉代替)(以Cl2計)消毒12h。
3)取2)中經消毒12h的水樣100mL滴定水中餘氯含量,然後向剩餘的0.9L水樣中加入過量5%的抗壞血酸(維生素C)溶液終止反應。再用H2SO4將pH調到0.5。對於每升酸化過的樣品加入90g Na2SO4使其飽和,然後再加入90mL甲基叔丁基醚萃取,放置15min後用45mL甲基叔丁基醚對上層有機層進行二次萃取,靜止5min後再將上層有機層轉移到旋轉蒸發儀中並且減壓濃縮到0.5mL,在加入10mL的乙腈旋轉蒸發到0.5mL。將上述經過酸化、加Na2SO4、多重萃取和旋蒸後的待測水樣用超純水稀釋至1mL,並用孔徑為0.45μm的聚四氟乙烯膜過濾。
4)採用本發明中的檢測方法同時對13種苯酚類氯代/溴代消毒副產物進行檢測。建立的超高效液相色譜/電噴霧電離三重四極杆質譜儀(UPLC/ESI-tqMS)檢測分析方法的參數設置如下:a.質譜參數:電離方式,電噴霧電離負離子模式;毛細管電壓,3.1kV;源溫度,130℃;脫溶劑溫度,500℃;脫溶劑氣流速,800L/h;錐孔氣流速,50L/h;13種化合物的多反應監測掃描方法所涉及的碰撞能、錐孔電壓和離子駐留時間見表1。b.液相色譜參數:進樣體積,5μL;色譜柱型號,Acquity UPLC HSS T3(2.1×100mm,填充物粒徑1.8μm,Waters);柱溫,30℃;流動相,水/甲醇;流速,0.4mL/min。梯度洗脫過程如下:在開始的8min裡,流動相水/甲醇的比例從95/5線性的變成5/95,在之後的0.1min裡面線性的變回95/5,這個組分繼續保持2.9min,使柱子平衡。
5)對步驟4)中得到的色譜圖採用已知物質保留值直接對照法、增加峰高法或三維圖譜檢測器定性法,初步判定樣品中的色譜峰是否與已知標準物質為同一物質。確定是同一種物質後再採用標準加入法分別計算樣品中的13種新型極性苯酚類氯代/溴代消毒副產物的濃度。
本實施例5中測定這13種新型極性苯酚類氯代/溴代消毒副產物所需總時間為11min。
本實施例5中氯胺消毒的實驗室模擬飲用水樣中,檢測到3,5-二氯-4-羥基苯甲醛、3,5-二溴-4-羥基苯甲醛、3,5-二氯-4-羥基苯甲酸、3,5-二溴-4-羥基苯甲酸、3,5-二氯水楊酸、3-溴-5-氯水楊酸、3,5-二溴水楊酸、2,4,6-三氯苯酚、2,6-二氯-4-溴苯酚、2,6-二溴-4-氯苯酚和2,4,6-三溴苯酚等11種待測消毒副產物,其中5種(3,5-二氯-4-羥基苯甲醛、3,5-二溴-4-羥基苯甲醛、3,5-二氯-4-羥基苯甲酸、3,5-二氯水楊酸和3,5-二溴水楊酸等)的濃度低於定量限,另外生成的6種待測消毒副產物濃度範圍為1.35-102.16ng/L。
氯消毒的實驗室模擬飲用水樣中,檢測到13種待測消毒副產物(見圖4),其中3,5-二氯-4-羥基苯甲酸消毒副產物的濃度低於定量限。另外生成的12種待測消毒副產物濃度範圍為2.64-241.1ng/L。
實施例6
本實施例6中,採用本發明的技術方案一次同時實現13種新型極性苯酚類氯代/溴代消毒副產物的分離與檢測。其具體方法包括如下步驟:
步驟1)配製不同濃度梯度的標準工作溶液,2)配置待測水樣,以及步驟3)待測水樣的預處理,與實施例1中所描述的方法相同。
步驟4)中,對步驟1)中所提及的13種苯酚類氯代/溴代消毒副產物進行一次同時檢測:建立的超高效液相色譜/電噴霧電離三重四極杆質譜儀(UPLC/ESI-tqMS)檢測分析方法的參數設置如下:a.質譜參數:電離方式,電噴霧電離負離子模式;毛細管電壓,2.5kV;源溫度,100℃;脫溶劑溫度,300℃;脫溶劑氣流速,600L/h;錐孔氣流速,25L/h;b.液相色譜參數:進樣體積,2μL;色譜柱型號,Acquity UPLC HSS T3(2.1×100mm,填充物粒徑1.8μm,Waters);柱溫,20℃;流動相,水/甲醇;流速,0.4mL/min。梯度洗脫過程如下:在開始的8min裡,流動相水/甲醇的比例從95/5線性的變成5/95,在之後的0.1min裡面線性的變回95/5,這個組分繼續保持2.9min,使柱子平衡。
步驟5)中,標準加入法校正曲線的繪製和結果計算:對步驟4)中得到的色譜圖採用已知物質保留值直接對照法、增加峰高法或三維圖譜檢測器定性法,初步判定樣品中的色譜峰是否與已知標準物質為同一物質。確定為同一種物質後再採用標準加入法分別計算樣品中的13種新型極性苯酚類氯代/溴代消毒副產物的濃度。
本實施例6中測定這13種新型極性苯酚類氯代/溴代消毒副產物所需總時間範圍為11min。
本實施例6中此方法的檢出限和定量限範圍分別為8.27-360.4ng/L和14.34-453.2ng/L。
實施例7
本實施例7中,採用本發明的技術方案一次同時實現13種新型極性苯酚類氯代/溴代消毒副產物的分離與檢測。其具體方法包括如下步驟:
步驟1)配製不同濃度梯度的標準工作溶液,2)配置待測水樣,以及步驟3)待測水樣的預處理,與實施例1中所描述的方法相同。
步驟4)中,對步驟1)中所提及的13種苯酚類氯代/溴代消毒副產物進行一次同時檢測:建立的超高效液相色譜/電噴霧電離三重四極杆質譜儀(UPLC/ESI-tqMS)檢測分析方法的參數設置如下:a.質譜參數:電離方式,電噴霧電離負離子模式;毛細管電壓,3.7kV;源溫度,150℃;脫溶劑溫度,650℃;脫溶劑氣流速,1000L/h;錐孔氣流速,200L/h;b.液相色譜參數:進樣體積,10μL;色譜柱型號,Acquity UPLC HSS T3(2.1×100mm,填充物粒徑1.8μm,Waters);柱溫,40℃;流動相,水/甲醇;流速,0.4mL/min。梯度洗脫過程如下:在開始的8min裡,流動相水/甲醇的比例從95/5線性的變成5/95,在之後的0.1min裡面線性的變回95/5,這個組分繼續保持2.9min,使柱子平衡。
步驟5)中,標準加入法校正曲線的繪製和結果計算:對步驟4)中得到的色譜圖採用已知物質保留值直接對照法、增加峰高法或三維圖譜檢測器定性法,初步判定樣品中的色譜峰是否與已知標準物質為同一物質。確定為同一種物質後再採用標準加入法分別計算樣品中的13種新型極性苯酚類氯代/溴代消毒副產物的濃度。
本實施例中測定這13種新型極性苯酚類氯代/溴代消毒副產物所需總時間範圍為11min。
本實施例中此方法的檢出限和定量限範圍分別為19.34-486.5ng/L和36.79-694.3ng/L。
本發明所述的檢測方法檢測限和定量限低,可滿足不同水體中這13種新型極性苯酚類氯代/溴代消毒副產物的準確檢測和定量,尤其是那些只含痕量的這些消毒副產物的水體;此外,該檢測方法可以對這13種新型極性苯酚類氯代/溴代消毒副產物進行很好的分離,實現一次同時檢測13種消毒副產物,大大提升了檢測效率。
以上顯示和描述了本發明的基本原理、主要特徵和本發明的優點。本行業的技術人員應該了解,本發明不受上述實施例的限制,上述實施例和說明書中描述的只是說明本發明的原理,在不脫離本發明精神和範圍的前提下,本發明還會有各種變化和改進,本發明要求保護範圍由所附的權利要求書、說明書及其等效物界定。