同軸靜電紡絲製備ICA‑SF/PLCL納米纖維膜的方法及作為GBR膜的應用與流程
2023-10-11 05:17:39 3
本發明涉及一種ica-sf/plcl複合納米纖維膜的製備,尤其涉及一種採用同軸靜電紡絲技術製備ica-sf/plcl納米纖維膜的方法;本發明同時還涉及該方法製備的ica-sf/plcl複合納米纖維膜作為gbr膜的應用,屬於複合材料技術領域和醫藥材料技術領域。
背景技術:
牙種植術目前已成為牙列缺損或缺失的首選修複方法;然而,由炎症(如肉芽組織)引起的骨缺損嚴重影響手術成功率。越來越多研究表明,引導骨再生(gbr)技術可有效恢復牙槽骨的高度和豐滿度,其原理是:通過阻礙成纖維細胞和上皮細胞長入骨缺損區而發揮屏障功能,並使成骨細胞附著、增殖增加,促進骨再生。
目前市場應用於gbr的材料,包括生物不可降解的聚四氟乙烯,該材料需二次手術取出,增加感染和膜暴露機率。因此,越來越多學者致力於生物可降解膜。bio-gide®是一種臨床常用的生物可降解膜,研究表明,其具有良好的生物相容性,低抗原性,足夠的機械性能和適益的降解性。為更好的誘導骨再生,複合性gbr膜(膜材料與骨誘導物質如生長因子相結合)已成為目前研究的熱點。
淫羊藿苷(icariin,ica,c33h40o15,分子量:676.67)是從中藥總黃酮中提取的主要活性成分,其具有多種重要的生理活性,如促進成骨細胞的增殖和分化以治療骨代謝疾病,及免疫調節和抗腫瘤活性。研究發現,ica可治療絕經後婦女的骨量喪失,通過增加opn、bmp表達促進骨髓間充質幹細胞分化成成骨細胞,最終恢復骨強度。同時,ica明顯抑制破骨細胞形成。因此認為,ica可以作為骨組織工程的成骨誘導劑。我們之前的研究發現,ica可以在體內外顯著促進人牙周膜幹細胞(hpdlscs)的增殖和成骨分化。
聚l-丙交酯(plla),具有良好的生物相容性和可降解性,使其更適宜在藥物控制釋放及組織工程等方面中應用。
絲素(sf)是一種天然結構蛋白,具有良好的生物學特性,如良好的細胞粘附性,可控降解率,機械強度,滲透性和抗酶降解性。它已經廣泛研究組織工程和控制藥物輸送系統。
同軸靜電紡絲是製備納米或亞微米纖維支架的簡單、有效方法,其在組織工程和藥物傳遞體系得到廣泛應用。其具有高的表面積/體積比,可以促進細胞附著,實現藥物卸載,並且達到持續和受控的藥物局部釋放。之前研究已證實,一些生物分子可成功包裹於納米纖維中,保持生物活性,纖維直徑可通過控制同軸靜電紡絲過程參數來控制。
因此,本發明採用同軸靜電紡絲技術擬製備一種ica-sf/plcl複合納米纖維膜,並對膜的理化性能、降解、釋藥及體內和外體對骨再生的作用進行研究,以期成為能持續釋放ica,促進骨再生功能的gbr膜。
技術實現要素:
本發明的目的是提供一種採用同軸靜電紡絲技術製備ica-sf/plcl複合納米纖維膜的方法;
本發明的另一目的是提供上述方法製備的ica-sf/plcl複合納米纖維膜作為誘導骨再生膜(gbr)的應用。
一、ica-sf/plcl複合納米纖維膜的製備
本發明ica-sf/plcl複合納米纖維膜的製備,包括以下工藝步驟:
(1)再生絲素蛋白的製備:將蠶繭浸於na2co3溶液中煮沸脫膠,並用蒸餾水洗滌除去絲膠,乾燥後在75~85℃下完全溶解於cacl2/ch3ch2oh/h2o的三元溶劑體系中,然後於蒸餾水中室溫下透析3~5d,過濾,冷凍乾燥,獲得再生絲素蛋白。
所述na2co3溶液的質量分數為0.2~0.5%;煮沸脫膠2次,每次0.5~1h;
所述cacl2/ch3ch2oh/h2o三元溶劑體系中,cacl2/ch3ch2oh/h2o的摩爾比為1/2/8。
(2)殼層紡絲液的製備:將上述所得再生絲素蛋白與plcl以1:2.3~1:2.5的質量比溶於六氟異丙醇中,且使再生絲素蛋白與plcl的總質量百分數為5~8%,常溫磁力攪拌5~6h,獲得殼層紡絲液。plcl的mw在1,000,000以上。
(3)芯層紡絲液的配製:將ica配製成濃度10-5μmol/l的水溶液作為芯層紡絲液;
(4)同軸靜電紡絲製備ica-sf/plcl複合納米纖維膜:將配製好的芯層紡絲液、殼層紡絲液分別裝入注射器中,連接同軸電紡雙噴頭,並與高壓靜電發生裝置相連接;控制同軸電紡噴頭與接收板間電壓為12~15kv,距離為12~15cm,芯層紡絲液、殼層紡絲溶液推進速度分別為0.1ml/h和1.0ml/h,在溫度25±2℃,相對溼度50±6%的環境下進行電紡,獲得電紡絲膜;再使電紡絲膜與戊二醛水溶液於密閉環境下交聯反應45~50h,真空乾燥,得ica-sf/plcl納米纖維膜樣品。
戊二醛溶液的質量百分數為25%。電紡絲膜的大小12mm×15mm,厚0.3~0.4mm時,戊二醛的用量為10ml。
二、ica-sf/plcl納米纖維膜表徵
1、掃描電鏡(sem)
將乾燥保存備用的樣本(3×3mm),真空噴金處理60s,應用jsm-4800型掃描電鏡(jeol,tokyo,japan)於10kv電壓條件下觀察。應用imagej圖像處理軟體(nationalinstitutesofhealth,md,usa)隨機選取纖維(n≧100),計算其平均直徑,並繪製纖維直徑分布圖。
ica-sf/plcl納米維膜sem圖像及纖維直徑分布圖如圖2顯示,見纖維表面形貌光滑連續,纖維直徑均一形成三維立體網狀結構,無串珠樣結構形成;纖維平均直徑為451.57±80.33nm,分布範圍為200~700nm,與天然細胞外基質結構相似,且納米級直徑纖維具有高的比表面積,可為細胞的粘附、生長提供理想的微環境,同時納米級纖維可攜載更多藥物。
2、透射電鏡(tem)
在紡絲過程中,用碳膜噴塗的銅網在電場中小心接取一定量的纖維絲,真空乾燥至有機溶劑充分揮發(約需7d)後,在100kv電壓條件下,應用feitecnaif3型透射電鏡(fei,usa)觀察纖維內部結構。
從ica-sf/plcl納米維膜的透射電鏡圖(圖3)可以看到,沿纖維長軸走向其內部形成明顯的芯殼結構。該芯殼結構形成的機理為:在同軸電紡過程中,殼層與芯層紡絲液在噴口處匯合,由於匯合時間短,加上聚合物擴散係數低,因此,芯殼層溶液固化前不會混合。同時,在高壓靜電場力作用下,芯殼層溶液的複合液滴表面聚集大量電荷,在電場力的作用下拉伸形成具有芯殼結構的納米纖維。其中,含原子數相對較多的ica在tem檢測過程中由於吸收電子相對較多,因此在tem圖像上顯示為相對較暗的纖維芯層,相對應的,sf/plcl顯示為纖維殼層,這暗示著中藥單體ica被成功包裹於纖維內部,形成了有效的藥物儲庫型緩釋系統。
3、x射線衍射(xrd)測試
製備的ica-sf/plcl納米纖維膜(15×15mm)採用d/max-2400型x射線多晶衍射儀(rigaku,tokyo,japan)對膜進行晶體結構的檢測。測試條件設置為:掃描速度為6°/min,功率為40kv/150ma,掃描範圍10~60°。
x射線衍射通過檢測材料衍射峰出現的位置,分析材料內部分子或原子結構以確定材料的結構特徵。以往的研究關於絲素蛋白和plcl構象表明,silkⅰ在x射線衍射測試中主要衍射峰接近12.2°、19.7°、24.7°和28.2°,plcl靜電紡絲膜在23.6°附近出現明顯的衍射峰。silkⅰ結構不穩定,構象規整性差;而silkii由於主要由結構穩定性較好的β摺疊構成,其構象規整性相對較好,穩定性好。ica-sf/plcl納米纖維膜的x射線衍射分析結果如圖4所示,圖像在17.2°左右出現新的衍射峰,而未檢測到其他明顯的衍射峰存在,說明芯層藥物ica的加入,對材料分子結構未產生明顯影響。電紡後的膜經2.5%的戊二醛交聯,絲素蛋白晶體結構的α螺旋轉變為穩定的β摺疊,結晶度增強,材料性能穩定性增加。
4、接觸角檢測
納米纖維膜(20×30mm)平整固定於親水角測試儀,吸取30μl去離子水小心低至膜表面,5s後,拍照並測量水滴切線與膜表面間的夾角,作為樣本接觸角。需在膜表面5個不同測試點分別測量,計算其平均值及標準差。
材料的潤溼性與細胞的粘附、增殖和遷移等行為密切相關。研究證實,純plcl纖維膜的接觸角為139.57°,其疏水性較強,表面不利於細胞的黏附、生長。而本研究的ica-sf/plcl載藥纖維膜的接觸角檢測結果(圖5)為125.72±4.93°,其親水性較純plcl纖維膜增加,因此,芯層藥物的加入可以增加膜的親水性,從而有利於細胞的黏附、生長。
5、拉伸強度測試
樣本(10×50mm)兩端寬10mm部分小心夾持於anscmt8102型電子萬能力學試驗機(hounsfield,uk)的夾具上,中間30mm作為測試區。檢測前,需用精密遊標卡尺測量樣本平均厚度。以10n測試力、1mm/min測試速度進行樣本的拉伸強度測試,並繪製相應的應力-應變曲線。
gbr膜需足夠的機械強度以抵抗周圍軟組織壓力,以維持有效的骨組織生長空間,避免缺損區塌陷,利於骨細胞的增殖分化。sf結構類似天然細胞外基質結構,為天然的細胞支架材料,但其力學性能較差;而plcl機械性能較好,將plcl與sf共混,可改善sf力學性能差的缺點。ica-sf/plcl納米纖維膜力學性能檢測結果(圖6)顯示,其拉伸強度為(5.54±0.46)mpa,力學性能良好,能滿足臨床gbr膜的強度要求。
6、體外釋藥行為檢測
將一定量的ica-sf/plcl納米纖維膜經紫外線正反面消毒各1h後,浸泡於30ml無菌pbs溶液,置於37℃、100rpm的恆溫水浴振蕩器震蕩,分別於測試的1、2、3、4、5、10、15、20、25、30d的同一時間點,取5ml釋放液,同時加入同體積的新鮮pbs溶液。從載藥膜釋放獲得的ica的量,通過應用uv2600型紫外分光光度計(shimadzu,japan)在λmax=270nm處檢測不同時間點收集的釋放液的吸光度值計算得到,繪製藥物的累積釋放曲線。
藥物緩釋系統以穩定速率將藥物釋放於靶位點,可提供局部有效藥物濃度,並避免全身藥物副作用。ica-sf/plcl載藥纖維膜體外釋藥結果如圖7所示,從該藥物累積釋放曲線可以看出,整個藥物釋放過程呈現兩個階段:初期的藥物快速釋放階段(最初的5d,藥物累積釋放率達47.54±0.06%)和之後的藥物穩定緩慢釋放階段(最終藥物累計釋放率為82.09±1.86%)。發生這兩個階段的其原因為:在藥物從材料釋放的初期,靠近載藥纖維表面的少量藥物首先快速釋放於周圍介質,之後由於材料在介質中的逐漸降解,纖維表面形成一些微孔,使得芯層藥物進一步以擴散的方式穩定、緩慢的釋放出來,從而可延長藥物從材料中釋放的時間,降低釋放速率,從而實現藥物的緩釋,達到發揮藥物最大功效的目的。
7、體外降解性能
樣本紫外線消毒,20ml無菌pbs(ph=7.4)為本研究的降解介質,置於37℃、100rpm的恆溫振蕩器連續震蕩,分別於測試的0、1、2、4、6周的同一時間點隨機取樣,蒸餾水衝洗乾淨,乾燥,用掃描電鏡及電子萬能力學試驗機分別對ica-sf/plcl納米纖維膜表面形貌及力學性能變化進行觀察和檢測。
理想的可植入材料的降解應與再生組織的生成同步發生。ica-sf/plcl載藥纖維膜體外降解結果如圖8所示。掃描電鏡圖片觀察發現,降解開始前,膜纖維表面連續光滑,之後由於降解介質的衝刷作用及芯層藥物的不斷釋放,纖維表面開始變得粗糙,出現明顯的蠶蝕狀空洞,但纖維膜整體結構尚存。同時,從每個時間點相對應的纖維膜的應力-應變曲線(圖9)可以看出,隨著降解現象的存在,ica-sf/plcl載藥纖維膜中的部分纖維斷裂,膜整體力學性能降低。說明該載藥纖維膜具有生物可降解性。
三、體外實驗
1、bmmscs的分離、培養與鑑定
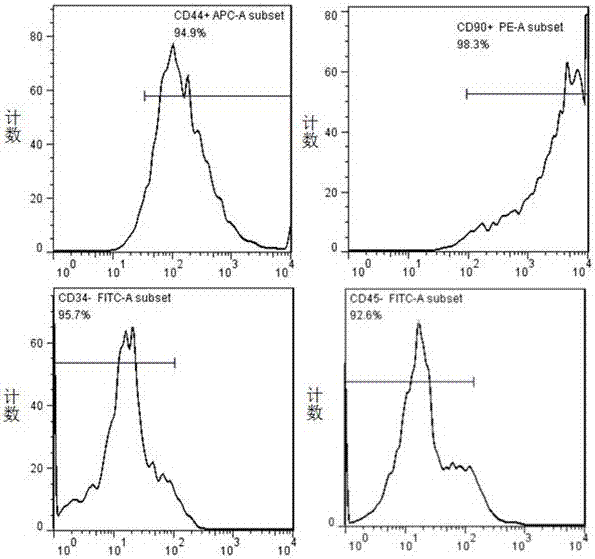
從蘭州大學醫學實驗中心購買3~4周齡雄性生理狀況良好的sd大鼠,採用過量麻藥(水合氯醛)處死,立即浸泡於盛有體積分數為75%酒精的容器中10min,置於無菌操作臺的手術板。嚴格無菌條件下獲得股骨,用含10%雙抗的無菌pbs衝洗3~5遍,剪去骨骺端,5ml注射器吸取含1%雙抗(gibco)、10%fbs的l-dmem(hyclone,usa)反覆衝洗骨髓腔至骨質發白,加入一定量完全培養液,將衝出的骨髓吹打混勻成單細胞懸液,於5%co2、37°c恆溫孵箱培養。24h後半量換液,之後每2~3天換液。細胞融合度達80%~90%時傳代培養。取第3代細胞進行以下相關實驗的研究。應用流式細胞儀(bdfacscanto,usa)檢測細胞表面標記,進行細胞鑑定:取p3代生長狀態良好的bmmscs,用0.25%胰酶充分消化細胞,1000rpm離心5min,用含1%bsa的pbs清洗細胞3~5次,細胞計數,依次加入單克隆抗體cd34、cd44、cd45和cd90。同時每管設立陰性對照。避光冰上孵育45min,用含1%bsa的pbs再次清洗細胞3~5次,清除未結合抗體,接著,用500μl含1%bsa的pbs將細胞重懸,流式細胞儀進行檢測分析。
流式細胞儀檢測結果如附圖1所示,分離培養的p3代bmmscscd44,cd90陽性表達率較高,陽性率分別為94.9%和98.3%,而cd34,cd45陽性率很低,分別為0.1%和0.7%,綜合以上數據,這些細胞是來自骨髓的bmmscs。
2、膜體外成骨性能檢測
將製備的ica-sf/plcl納米纖維膜按100g/l的比例浸泡於含10%胎牛血清的l-dmem培養基內,37℃、5%co2條件下連續浸提72h,離心過濾後獲得浸提液。備用。
生長狀態良好的p3代bmmscs以1.0×104cells/ml的密度接種於細胞培養皿,加入10ml完全培養液,37℃、5%co2孵箱中孵育;24h後,實驗組加入浸提液培養,對照組用普通完全培養液培養,每三天換液。用2%茜素紅染液(genmedscientificsinc.,usa)進行鈣結節檢測。細胞培養14d後,4%多聚甲醛固定,37℃條件下,茜素紅染色30min檢測鈣結節。
生長狀態良好的p3代bmmscs胰酶消化後,按1×104個/ml接種於3塊96孔板上,37℃、5%co2孵箱中孵育24h後,棄去舊培養液。實驗分為2組,每組設6個復孔,實驗組加入浸提液,對照組加入普通完全培養液,每3天換液一次。分別於第3、7、14d取一塊96孔板,棄培養液,pbs清洗3遍,每孔加入500μl1%的tritonx-100(ambion,bylifetechnologies),37℃培養箱裂解1h,收集細胞裂解液,按鹼性磷酸酶試劑盒(南京建成生物工程研究所,南京,中國)說明依次加入各試劑,酶聯免疫檢測儀(elx800,bio-tek,winooski,vt,usa)於520nm處測定各孔alp值,以均值進行統計分析(應用spss17.0軟體進行統計學分析,數據以平均值±標準偏差表示。通過單因素方差分析(anova)評價各組間的統計學差異;p0.05),無膜覆蓋組幾乎觀察不到新骨形成。術後8周,所有組均有新骨形成,與sf/plcl組及無膜覆蓋組比較,ica-sf/plcl納米纖維膜組骨缺損癒合顯著較快,新骨體積和密度分別為6.09±1.27mm3、6.43±0.17%。sf/plcl組與無膜覆蓋組新骨生成體積差異無統計學意義(p>0.05)。術後12周,雖然所有組均顯示有新骨生成,但在ica-sf/plcl納米纖維膜組,新生骨已擴展覆蓋大部分缺陷區域,新骨體積和密度分別為約15.95±3.58mm3和14.02±0.93%,顯示最快和最多骨形成。而sf/plcl組和無膜覆蓋組的新骨形成僅局限於缺損中心或在缺損邊緣。因此,我們證實了從ica-sf/plcl納米纖維膜釋放的ica的具有體內成骨分化活性。
3、組織學檢測
固定後的樣本經脫鈣複合液(甲酸12ml、鹽酸8ml、甲醛5ml、蒸餾水75ml)脫鈣、梯度酒精脫水後,常規石蠟包埋,切片,蘇木精和伊紅(nanjingjianchengbiotechnologyinstitute)染色,光鏡(olympus,ix70,japan)下觀察。使用imagej軟體(nationalinstitutesofhealth,md,usa)定量分析新骨形成,並計算新骨面積與總缺損面積比。
術後4、8、12周樣本進行組織學分析進一步表徵新骨形成,並計算新骨面積與總缺損面積的比率(圖13)。術後4周,ica-sf/plcl納米纖維膜組和sf/plcl組均可觀察到有新骨生成,但無膜覆蓋組其骨缺損區被大量纖維結締組織填充,幾乎無新骨生成。術後8周,ica-sf/plcl納米纖維膜組顯示比sf/plcl組或無膜覆蓋組更多的新骨形成。其中無膜覆蓋組可觀察到大量纖維結締組織。術後12周,ica-sf/plcl納米纖維膜組新骨面積顯著高於其他組。sf/plcl組是與纖維脂肪組織混合的新骨填充骨缺陷,無膜覆蓋組只有一些纖維結締組織。
綜上所述,我們利用同軸靜電紡絲成功製備了ica-sf/plcl納米纖維膜,骨促進生長因子ica可包裹於纖維中而無結構完整性或功能的變化。體外成骨誘導分化和體內應用於骨缺損實驗表明,該載藥膜可持續、有效地釋放ica而顯著促進骨生成,且無細胞毒性。因此,應用同軸電紡技術製備的ica-sf/plcl納米纖維膜是一種有前景的gbr膜。
附圖說明
圖1為大鼠骨髓間充質幹細胞表面標誌物的表達。
圖2為ica-sf/plcl載藥纖維膜sem圖(a:×5000,b:×10000)和纖維直徑分布圖(c)。
圖3為ica-sf/plcl載藥纖維tem圖(×50000)。
圖4為ica-sf/plcl載藥纖維膜xrd圖。
圖5為ica–sf/plcl載藥纖維膜的親水角。
圖6為ica–sf/plcl納米纖維膜應力-應變曲線。
圖7為ica–sf/plcl納米纖維膜體外藥物釋放曲線。
圖8為ica–sf/plcl載藥纖維膜表面形態(a~e)註:a:降解0周;b:降解1周;c:降解2周;d:降解4周;e:降解6周。
圖9為ica–sf/plcl載藥纖維膜拉伸強度(a~e)變化。註:a:降解0周;b:降解1周;c:降解2周;d:降解4周;e:降解6周。
圖10為bmmscs培養於ica-sf/plcl納米纖維膜浸提液後的茜素紅染色結果(a)及alp分(b)。
圖11為觀察bmscs與ica-sf/plcl載藥纖維膜複合後的細胞形態的sem(左:×1500;右:×3500)
圖12為bmscs在ica-sf/plcl載藥纖維膜上培養不同時間增殖情況。註:ica代表ica–sf/plcl載藥纖維膜組。
圖13為顱骨缺損的μ-ct圖像。
圖14為定量分析骨缺損區的新骨體積(a)j及密度百分比(b)。*與對照相比,骨形成明顯增多(註:*p<0.05,**p<0.01。sf表示sf/plcl膜組;ica表示ica-sf/plcl納米纖維膜組)。
圖15為顱骨缺損的新骨形成的組織學分析。(a)顱骨缺損的he染色圖像。比例尺=0.2mm。(b)定量分析顱骨缺損中新骨面積百分比。
具體實施方式
下面通過具體實施例對本發明採用同軸靜電紡絲技術製備ica-sf/plcl納米纖維膜的方法作進一步的說明。
(1)再生絲素蛋白的製備:將蠶繭(浙江桐廬,中國)浸於質量分數0.5%的na2co3溶液中煮沸脫膠2次,每次1h;並用蒸餾水洗滌3此除去絲膠,50℃乾燥12h後,再在80℃下完全溶解於cacl2/ch3ch2oh/h2o的三元溶劑體系中(cacl2/ch3ch2oh/h2o的摩爾比為1/2/8),然後於蒸餾水中室溫下透析4d,過濾,冷凍乾燥,獲得再生絲素蛋白sf。
(2)殼層紡絲液的製備:將上述所得sf與plcl以1:2.3~1:2.5的質量比溶於六氟異丙醇中,且使sf與plcl(mw=1,000,000;naramedicaluniversity,japan)的總質量百分數為8%,常溫磁力攪拌6h,獲得殼層紡絲液;
(3)芯層紡絲液的配製:將ica配製成濃度10-5μmol/l的水溶液作為芯層紡絲液;
(4)同軸靜電紡絲製備ica-sf/plcl複合納米纖維膜:將配製好的芯層紡絲液、殼層紡絲液分別裝入10ml注射器中,連接同軸電紡雙噴頭,並與高壓靜電發生裝置(dw-p503-1acdf;北京,中國)相連接;控制同軸電紡噴頭與接收板間電壓為15kv,距離為15cm,芯層紡絲液、殼層紡絲溶液推進速度分別為0.1ml/h和1.0ml/h,在溫度25℃左右,相對溼度50±6%的環境下進行電紡,獲得電紡絲膜;再使電紡絲膜用25%的戊二醛(國藥化學試劑有限公司,中國)(電紡絲膜的大小12mm×15mm,厚0.3~0.4mm時,戊二醛的用量10ml)作為交聯劑置於密閉玻璃乾燥皿內交聯48h,真空乾燥,即得ica-sf/plcl納米纖維膜樣品。