用於有機電致發光元件的化合物及使用該化合物的有機電致發光元件的製作方法
2023-09-18 01:54:46 4
本發明有關一種用於有機電致發光元件的化合物及使用該化合物的有機電致發光元件。
背景技術:
為滿足有機電致發光元件(oled)的應用,已更重視新穎有機材料的開發。這些元件因為在製造高發光率的高密度像素顯示器上提供有成本優勢,更具有長壽命、高效率、低驅動電壓及廣色域,而具商業性吸引力。
典型的oled包括至少一夾置於陽極與陰極之間的有機發射(organicemissive)層。當施加電流時,陽極注入空穴且陰極注入電子至該一層或多層有機發射層,被注入的空穴及電子各自遷移(migrate)至相反的帶電荷電極。當電子及空穴局限在相同的分子上時,形成「激子(exciton)」,該激子具有激發能態的局限化電子-空穴對。激子通過發光機制鬆弛而發射光。為了提升此等元件的電荷傳輸能力及發光效率,已於發光層旁結合一層或多層的額外層體,例如電子傳輸層及/或空穴傳輸層,或電子阻擋層及/或空穴阻擋層。us5707745及us9153787已揭示在主體材料摻雜另一材料(客體),以提升元件性能及調整色度。
製造具多層薄膜結構oled的原因包含使這些電極及有機層之間的界面安定。此外,在有機材料中,電子及空穴的遷移率(mobility)明顯不同。因此,若使用相稱的空穴傳輸及電子傳輸層,空穴及電子可有效地傳輸至發光層。此外,若發射層中空穴及電子的密度平衡時,可增加發光效率。適當的結合上述有機層可增進元件效率及壽命。然而,仍難以找到滿足所有實際顯示器應用需求的有機材料。
因此,亟需開發一種電子傳輸材料,以延長元件壽命,並可提高發光效率和維持低的驅動電壓。
技術實現要素:
本發明一目的在於提供一種具有高發光效率、低驅動電壓及較長壽命的用於oled的材料。
本發明提供一種用於oled的式(i)化合物:
其中,
x1表示經取代或未經取代的(c6-c30)芳基、經取代或未經取代的(5至30元)雜芳基;
a1表示經取代或未經取代的(c6-c30)芳基、經取代或未經取代的(5至30元)雜芳基;
x1、及a1為相同或相異;以及
n表示1或2的整數,當n表示2時,a1各自為相同或相異;且當x1為經取代或未經取代的(c6-c30)芳基時,n為1。
本發明還提供一種有機電致發光元件,包含:陰極;陽極;以及有機層,介於陰極與陽極之間,且該有機層包含上述式(i)化合物。
本發明的有機電致發光元件的有機層可為電子傳輸層、電子注入層、發光層、空穴阻擋層或電子阻擋層,且除了該有機層外,該有機電致發光元件還可包括不同於該有機層的電子傳輸層、電子注入層、發光層、空穴阻擋層及電子阻擋層所組成組的至少一層。
根據本發明,藉由本發明提供的式(i)化合物,可改善元件的穩定性,並降低操作電壓。
附圖說明
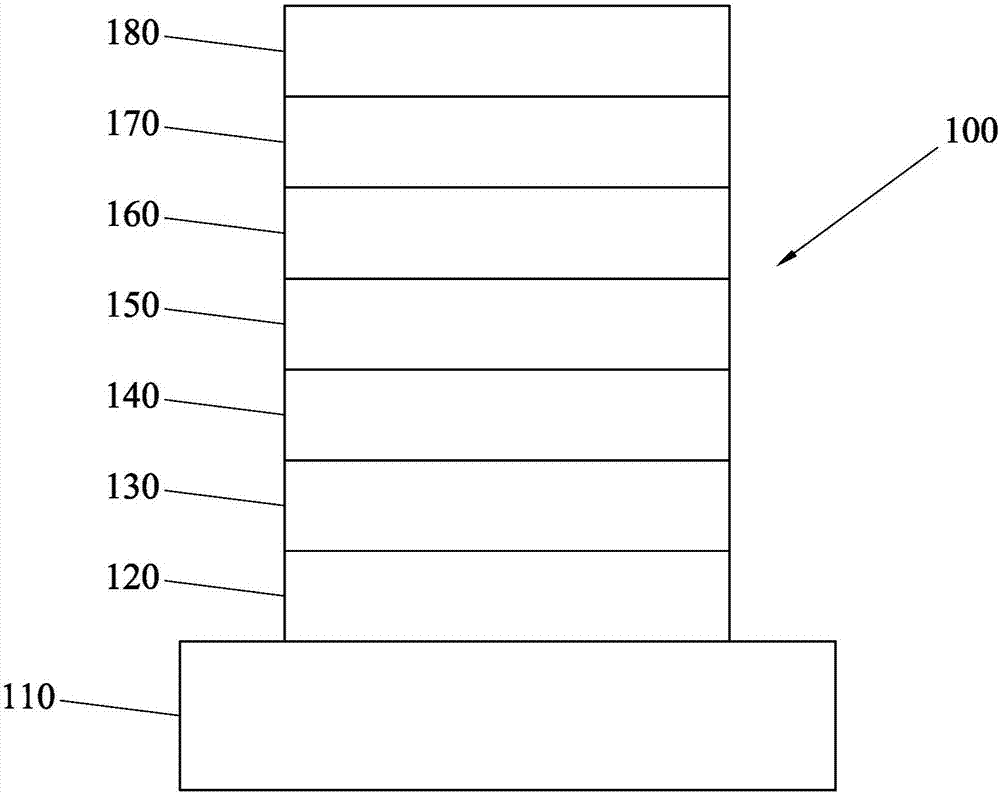
圖1為本發明有機電致發光元件的一實施例的剖面示意圖;
圖2為本發明有機電致發光元件的另一實施例的剖面示意圖;
圖3為本發明有機電致發光元件的又一實施例的剖面示意圖;以及
圖4為本發明有機電致發光元件的電致發光光譜。
符號說明
100、200、300有機電致發光元件
110、210、310基底
120、220、320陽極
130、230、330空穴注入層
140、240、340空穴傳輸層
150、250、350發光層
160、260、360電子傳輸層
170、270、370電子注入層
180、280、380陰極
245、355激子阻擋層。
具體實施方式
以下藉由較佳實施例詳細說明本發明,以使本領域技術人員易於了解本發明的說明書所揭示的益處及功效。
本文所述的範圍與所揭露的值都包含邊值且可合併。舉例而言,當任何數值,例如一整數或點落入本文所述的範圍時,則可以該點或數值做為下限或上限值而推導出次範圍。此外,本文所例舉基團,例如屬於x1和a1的基團或取代基,皆可與其他基團合併於式(i)。
依據本發明,可應用於oled的化合物如式(i)所示:
其中,x1表示經取代或未經取代的(c6-c30)芳基、經取代或未經取代的(5至30元)雜芳基;
a1表示經取代或未經取代的(c6-c30)芳基、經取代或未經取代的(5至30元)雜芳基;
x1及a1為相同或相異;以及
n表示1或2的整數,當n表示2時,a1各自為相同或相異;且當x1為經取代或未經取代的(c6-c30)芳基時,n為1。
於一具體實施例中,x1表示經取代或未經取代的(c6-c20)芳基、經取代或未經取代的(5至20元)雜芳基,該(5至20元)雜芳基含有至少一個選自由n、o及s所組成組中的雜原子。
於一具體實施例中,a1表示經取代或未經取代的(c6-c20)芳基、經取代或未經取代的(5至20元)雜芳基,該(5至20元)雜芳基含有至少一個選自由n、o及s所組成組中的雜原子。
於一具體實施例中,上述式(i)中的a1表示式(ii)或式(iii)化合物:
式(ii)中,ar1及ar2各自獨立表示未經取代的(c6-c20)(亞)芳基,式(ii)化合物藉由ar1或ar2與式(i)化合物連接,或式(ii)化合物藉由ar1及ar2與式(i)化合物形成稠合環;式(iii)中,z1表示n、o、s、cme2或ch2,其中,當z1為n時,式(iii)化合物藉由z1與式(i)化合物連接,且當z1為o、s、cme2或ch2時,式(iii)化合物藉由其所具的苯基與式(i)化合物連接。
於一具體實施例中,當n為1時,該式(i)化合物為式(i-1)或式(i-2)所示:
於一具體實施例中,當n為2時,該式(i)化合物為式(i-3)所示:
文中,表達成「經取代或未經取代的」中的「經取代的」表示在某個官能團中的氫原子經另一個原子或基團(即取代基)置換。這些取代基各自獨立為選自下列所組成組中的至少一者:氘;滷素;(c1-c30)烷基;(c1-c30)烷氧基;(c6-c30)芳基;(5至30元)雜芳基,其中該(5至30元)雜芳基可經(c6-c30)芳基取代;經(c6-c30)芳基取代的(5至30元)雜芳基;(c3-c30)環烷基;(5至7元)雜環烷基;三(c1-c30)烷基矽烷基;三(c1-c30)芳基矽烷基;二(c1-c30)烷基(c6-c30)芳基矽烷基;(c1-c30)烷基二(c6-c30)芳基矽烷基;(c2-c30)烯基;(c2-c30)炔基;氰基;二(c1-c30)烷基胺基;二(c6-c30)芳基胺基;(c1-c30)烷基(c6-c30)芳基胺基;二(c6-c30)芳基硼基;二(c1-c30)烷基硼基;(c1-c30)烷基(c6-c30)芳基硼基;(c6-c30)芳基(c1-c30)烷基;(c1-c30)烷基(c6-c30)芳基;羧基;硝基;及羥基。
於一具體實施例中,該(c6-c30)芳基經選自(c1-c10)烷基、(c6-c30)芳基及(5至30元)雜芳基所組成組中的至少一種取代基取代,其中,該(5至30元)雜芳基經(c6-c30)芳基取代。
於一具體實施例中,該(5至30元)雜芳基經選自(c1-c10)烷基、(c6-c30)芳基及(5至30元)雜芳基所組成組中的至少一種取代基取代,其中,該(5至30元)雜芳基經(c6-c30)芳基取代。
於另一具體實施例中,該(c6-c30)芳基經選自(c1-c5)烷基、(c6-c10)芳基及苯並咪唑基所組成組中的至少一種取代基取代,其中,該苯並咪唑基經(c6-c10)芳基取代。
於另一具體實施例中,該(5至30元)雜芳基經選自(c1-c5)烷基、(c6-c10)芳基及苯並咪唑基所組成組中的至少一種取代基取代,其中,該苯並咪唑基經(c6-c10)芳基取代。
文中,「(亞)芳基」表示芳基或亞芳基。芳基指衍生自芳香烴的單環或稠環,且包括苯基、聯苯基、聯三苯基(terphenyl)、萘基、聯萘基、苯基萘基、萘基苯基、芴基(fluorenyl)、苯基芴基、苯並芴基、二苯並芴基、菲基、苯基菲基、蒽基、茚基、聯伸三苯基(triphenylenyl)、芘基、稠四苯基、苝基、蒯基(chrysenyl)、萘並萘基、丙二烯合芴基(fluoranthenyl)等。
文中,「(亞)雜芳基」表示雜芳基或亞雜芳基。(5至30元)雜芳基指含有選自b、n、o、s、p(=o)、si、及p所組成組中的至少一個雜原子且具有3至30個環主鏈原子的芳基;為單環,或為與至少一個苯環縮合的稠環;為部分飽和;為通過一或多個單鍵連結至少一個雜芳基或芳基至雜芳基所形成者;及包括單環型雜芳基如,呋喃基、噻吩基、吡咯基、咪唑基、吡唑基、噻唑基、噻二唑基、異噻唑基、異噁唑基、噁唑基、噁二唑基、三嗪基、四嗪基、三唑基、四唑基、呋呫基、吡啶基、吡嗪基、嘧啶基、噠嗪基等,及稠環型雜芳基如,苯並呋喃基、苯並噻吩基、異苯並呋喃基、二苯並呋喃基、二苯並噻吩基、苯並咪唑基、苯並噻唑基、苯並異噻唑基、苯並異噁唑基、苯並噁唑基、異吲哚基、吲哚基、吲唑基、苯並噻二唑基、喹啉基、異喹啉基、噌啉基、喹唑啉基、喹噁啉基、咔唑基、啡噁嗪基、啡啶基、苯並二噁茂基(benzodioxolyl)、二氫吖啶基等。
於一具體實施例中,x1選自下列所組成組中的一者:
及
於一具體實施例中,a1選自下列所組成組中的一者:
及
本發明還提供一種有機電致發光元件,包含:陰極;陽極;以及有機層,介於該陰極與陽極之間,且該有機層包含上述式(i)化合物。
前述式(i)化合物的優選實例選自但不限於下列1-1至1-10、2-1至2-10、3-1至3-10、4-1至4-10、5-1或2-10、6-1至6-10、7-1至7-10、8-1至8-10、9-1至9-10、10-1至10-10、11-1至11-10、12-1至12-10、13-1至13-10、14-1至14-10、15-1至15-10、16-1至16-10、17-1至17-10、18-1至18-10、19-1至19-10、20-1至20-10、21-1至21-10、22-1至22-10、23-1至23-10、24-1至24-10、25-1至25-10、26-1至26-10、27-1至27-10、28-1至28-10、29-1至29-10、30-1至30-10、31-1至31-10、32-1至32-10、33-1至33-10、34-1至34-11、35-1至35-11、36-1至36-11。
可採用如下所示反應路線1至6中所示的一系列反應以合成式(i)所示化合物,但不限於此。
將4-溴苯甲醛(4-bromobenzaldehyde)(50g,270.2mmol)、4-乙醯基聯苯(4-acetylbiphenyl)(55.68g,283.8mmol)及1000mletoh加入反應瓶中攪拌,再加nao-t-bu(sodiumtert-butoxide,簡稱stb)(29.83g,310.8mmol)至反應瓶中一起攪拌後,室溫25℃反應隔夜(通氮氣),過濾後以100ml水及100ml甲醇衝洗,固體再以400ml純水與100ml甲醇攪拌15分鐘後過濾重複2~3次,固體再以400ml甲醇攪拌15分鐘過濾,固體再以400ml乙醇攪拌15分鐘純化過濾,65℃烘乾得84.7g的白色固體中間體a,產率86.28%。
中間體a(20g,55.1mmol)與不同的氯化吡啶(pyridiniumchloride)衍生物(60.6mmol)及200ml甲苯入反應瓶中攪拌,再加入stb(7.93g,82.6mmol)至反應瓶中一起攪拌後,溫度100±5℃反應16小時(通氮氣)。降溫,加300ml乙酸乙酯(ethylacetate,簡稱ea)後,以200ml純水萃取4次,以無水硫酸鈉除水過濾,濃縮後加甲醇析出過濾,以甲醇衝洗烘乾得固體的中間體b。
將3-溴苯甲醛(3-bromobenzaldehyde)(50g,2702mmol)、4-乙醯基聯苯(4-acetylbiphenyl)(55.68g,2838mmol)及1000mletoh加入反應瓶中攪拌,再加stb(29.83g,3108mmol)至反應瓶中一起攪拌後,室溫25℃反應隔夜(通氮氣),過濾後以100ml水跟100ml甲醇衝洗,固體再以400ml純水與100m甲醇攪拌15分鐘後過濾重複2~3次,固體再以400ml甲醇攪拌15分鐘過濾,固體再以400ml乙醇攪拌15分鐘純化過濾,65℃烘乾得89.6g的白色固體中間體c,產率91.27%。
中間體c(20g,551mmol)與不同的氯化吡啶衍生物(606mmol)與200ml甲苯入反應瓶中攪拌,再加入stb(7.93g,826mmol)至反應瓶中一起攪拌後,溫度100±5℃反應16小時(通氮氣)。降溫加300mlea後以200ml純水萃取4次,以無水硫酸鈉除水過濾,濃縮後加甲醇析出過濾,以甲醇衝洗烘乾得固體的中間體d。
將3,5-二溴苯甲醛(3,5-dibromobenzaldehyde)(30g,1137mmol)、4-乙醯基聯苯(4-acetylbiphenyl)(23.42g,1194mmol)及600mletoh加入反應瓶中攪拌,再加stb(16.37g,1705mmol)至反應瓶中一起攪拌後,室溫25℃反應隔夜(通氮氣),過濾後以100ml水跟100ml甲醇衝洗,固體再以250ml純水與50m甲醇攪拌15分鐘後過濾重複2~3次,固體再以200ml甲醇攪拌15分鐘過濾,固體再以250ml乙醇攪拌15分鐘純化過濾,65℃烘乾得48.5g的淡黃色固體中間體e,產率98.49%。
中間體e(20g,551mmol)與不同的氯化吡啶衍生物(606mmol)與200ml甲苯入反應瓶中攪拌,再加入stb(7.93g,826mmol)至反應瓶中一起攪拌後,溫度100±5℃反應16小時,通氮氣。降溫加300mlea後以200ml純水萃取4次,以無水硫酸鈉除水過濾,濃縮後加甲醇析出過濾,以甲醇衝洗烘乾得固體的中間體f。
反應路線1
反應路線2
反應路線3
反應路線4
反應路線5
反應路線6
本發明還提供一種有機電致發光元件,包含:陰極;陽極;以及包含本發明式(i)化合物的有機層,介於該陰極與陽極之間。
式(i)所示化合物可應用於oled的有機層。因此,本發明的oled具有至少一層設置於基底上陽極與陰極之間的有機層,其中有機層包含如上列式(i)所示化合物。該有機層為發光層、空穴阻擋層、電子傳輸層、電子注入層或空穴傳輸層,且除了該有機層外,該有機電致發光元件還可包括不同於該有機層的電子傳輸層、電子注入層、發光層、空穴阻擋層及電子阻擋層所組成群組的至少一層。包含式(i)所示化合物的該有機層優選為電子傳輸/注入層,可以式(i)所示化合物作為單一材料,或與n/p型導電摻雜劑(n/ptypedopants)結合。於一具體實施例中,該電子傳輸層包含式(i)化合物。
應用於電子傳輸層的導電摻雜劑優選為有機鹼金屬/鹼土金屬絡合物(organicalkali/alkalinemetalcomplexes)、氧化物(oxides)、滷化物(halides)、碳酸鹽(carbonates)及含有至少一種選自鋰和銫金屬的磷酸鹼金屬/鹼土金屬鹽(phosphatesofalkali/alkalinegroupmetalscontainingatleastonemetalselectedfromlithiumandcesium)。該有機金屬絡合物在上述專利或他處為已知,並可選擇合適的有機金屬絡合物於本發明中使用。
以該有機層的重量計算,該導電摻雜劑的含量為0wt%至90wt%。例如,該有機層為電子傳輸層或電子注入層,前述導電摻雜劑於電子傳輸層/電子注入層中的含量優選25wt%至75wt%重量比的範圍。
於一具體實施例中,該有機層是電子傳輸層。
於一具體實施例中,該電子傳輸層包含式(i)化合物與n型導電摻雜劑,且該n型導電摻雜劑的含量為大於0wt%至75wt%。
於一具體實施例中,以該有機層的重量計算,式(i)化合物的含量為25wt%至100wt%。又,該有機層的厚度為1納米至500納米。
於又一具體實施例中,該有機電致發光元件還包括電子傳輸層、電子注入層、發光層、空穴阻擋層及電子阻擋層所組成組的至少一層。而該發光層還包含螢光或磷光發射體。
於一具體實施例中,自該陽極至該有機層之間還包括空穴注入層、空穴傳輸層、發光層,且自該有機層至該陰極之間還包括電子注入層,且該有機層為電子傳輸層。
再者,式(i)所示化合物還可被用於發光層與電子傳輸層間的層。該發光層可包含螢光及磷光摻雜劑以及分別對應螢光或磷光摻雜劑的螢光及磷光發射主體。
進一步而言,由下式(i)所示化合物可被使用於電子注入/傳輸層或空穴阻擋層及/或電子阻擋層。
本發明的有機電致發光元件的結構將配合圖式加以說明。
圖1為本發明有機電致發光元件的一具體實施例的剖面示意圖。有機電致發光元件100包含基底110、陽極120、空穴注入層130、空穴傳輸層140、發光層150、電子傳輸層160、電子注入層170及陰極180。有機電致發光元件100可經由依序沉積上述各層來製作。圖2為本發明有機電致發光元件另一具體實施例的剖面示意圖。圖2所示的有機電致發光元件與圖1近似,除了包含基底210、陽極220、空穴注入層230、空穴傳輸層240、發光層250、電子傳輸層260、電子注入層270及陰極280,其不同之處在於激子阻擋層245設於空穴傳輸層240與發光層250之間。圖3繪示本發明有機電致發光元件又一具體實施例的剖面示意圖。圖3所示的有機電致發光元件亦與圖1近似,除了包含基底310、陽極320、空穴注入層330、空穴傳輸層340、發光層350、電子傳輸層360、電子注入層370及陰極380,其不同之處在於激子阻擋層355設於發光層350與電子傳輸層360之間。
亦可依圖1至3所示元件的反置式結構(reversestructure)製造有機電致發光元件。於反置式結構中,可視需求增減一層或數層。
應用於空穴注入層、空穴傳輸層、電子阻擋層、空穴阻擋層、發光層及電子注入層的材料可選擇常用材料。例如,形成電子傳輸層的電子傳輸材料不同於形成發光層的材料,且其具有傳輸空穴的性質,從而促成空穴於電子傳輸層中遷移,且防止因發光層與電子傳輸層的解離能差所導致的載子累積。
此外,us5844363揭示一種結合陽極的可撓性透明基底,其全部內容為本發明所引用。如us20030230980所例示p型摻雜的空穴傳輸層以摩爾比50:1於m-mtdata摻雜f4-tcnq,其全部內容為本發明所引用。如us20030230980所例示n型摻雜的電子傳輸層以摩爾比1:1於bphen摻雜鋰,其全部內容為本發明所引用。如us5703436及us5707745所例示陰極的全部內容為本發明所引用,該陰極具有金屬薄層,如:鎂/銀(mg:ag),及以濺射沉積覆蓋金屬薄層的透明導電層(itolayer)。us6097147及us20030230980所揭示各阻擋層的應用及原理,其全部內容為本發明所引用。us20040174116所例示的注入層及同案所說明的保護層,其全部內容為本發明所引用。
未特別說明的結構及材料亦可應用於本發明,如us5247190所揭示包括聚合物材料(pleds)的有機電致發光元件,其全部內容為本發明所引用。再者,具有單一有機層的有機電致發光元件或如us5707745所揭示堆棧形成的有機電致發光元件,其全部內容為本發明所引用。
除有特別限定,不同實施例中的任何層可使用任何適當方法來沉積形成。以有機層而言,較佳的方法包含如us6013982及us6087196所揭示的熱蒸鍍法及噴印法,其全部內容為本發明所引用;us6337102所揭示有機氣相沉積法(organicvaporphasedeposition,ovpd),其全部內容為本發明所引用;us10/233470所揭示有機氣相噴印沉積法(depositionbyorganicvaporjetprinting,ovjp),其全部內容為本發明所引用。其他適當方法包含旋轉塗布及以溶液為基礎的製程。以溶液為基礎的製程優選在氮氣或惰性氣體環境中進行。對於其他層而言,較佳的方法包含熱蒸鍍法。較佳的圖案化方法包含如us6294398及us6468819所揭示通過遮罩沉積再冷焊的製程,及整合噴印或有機氣相噴印沉積與圖案化的製程,其全部內容為本發明所引用。當然亦可使用其他方法。可調整用於沉積的材料以對應其所特用的沉積方法。
本發明式(i)所示化合物能以真空沉積或旋轉塗布法製成應用於有機電致發光元件的非晶薄膜。當該化合物使用於任一上述有機層,其以高發光效率及低驅動電壓展現出較長使用壽命及較佳熱穩定性。
本發明的有機電致發光元件可應用於單一元件,其結構為陣列配置或陣列x-y坐標中設有陰陽兩極的元件。相較於習知元件,本發明能顯著提升有機電致發光元件的發光效率及驅動穩定性。此外,與發光層中的磷光摻雜劑相結合,本發明的有機電致發光元件應用於全彩或多彩顯示面板能實現較佳性能且可發射白光。
以下藉由實施例詳細說明本發明的諸多性質及功效。該詳述實施例僅用於說明本發明的性質,本發明不限於特定實施例所例示者。
合成例1(化合物1-4的合成)
將4-二苯基-4-基-6-(4-溴苯基)-2-吡啶-3-基-嘧啶(4-biphenyl-4-yl-6-(4-bromophenyl)-2-pyridin-3-yl-pyrimidine)(10g,215mmol)與3-(3-吡啶基)苯基硼酸(3-(3-pyridyl)phenylboronicacid)(5.14g,258mmol)放入反應瓶中,再加入甲苯100毫升,再將k2co3(4.46g,323mmol)溶解於40毫升的去離子水中加入反應瓶內,加入pd(pph3)4(0.371g,0.3mmol)再加入酒精20毫升,攪拌及於80℃回流反應16小時。反應完後加入150毫升去離子水攪拌至室溫,過濾出固體並以去離子水跟丙酮清洗,該固體再加入200毫升去離子水、50毫升甲醇及50毫升丙酮的混合溶液中攪拌30分鐘,再過濾,重複2次。烘乾該固體,再以1000ml甲苯加熱溶解後,通過矽膠柱,蒸餾濃縮後加入300ml甲醇攪拌30分鐘過濾,烘乾得白色固體的化合物1-4(4.7g,產率40.5%)。
1hnmr(400mhz,cdcl3,δ):
9.943(s,1h);9.001-8.981(d,h);8.944(s,1h);8.776-8.763(d,1h);8.647-8.640(d,1h);8.436-8.391(t,4h);8.167(s,1h);7.982-7.961(d,1h);7.878-7.813(m,5h);7.741-7.633(m,5h);7.526-7.419(m,5h)。
合成例2(化合物1-5的合成)
將4-二苯基-4-基-6-(4-溴苯基)-2-吡啶-3-基-嘧啶(4-biphenyl-4-yl-6-(4-bromophenyl)-2-pyridin-3-yl-pyrimidine)(10g,215mmol)與3-二苯基硼酸(3-biphenylboronicacid)(5.12g,258mmol)放入反應瓶中,再加入甲苯100毫升,再將k2co3(4.46g,323mmol)溶解於40毫升的去離子水中加入反應瓶內,加入pd(pph3)4(0.371g,0.3mmol)再加入酒精20毫升,攪拌及於80℃回流反應16小時。反應完後加入150毫升去離子水攪拌至室溫,過濾出固體並以去離子水跟丙酮清洗,該固體再加入200毫升去離子水、50毫升甲醇及50毫升丙酮的混合溶液中攪拌30分鐘,再過濾,重複2次。烘乾該固體,再以1000ml甲苯加熱溶解後,通過矽膠柱,蒸餾濃縮後加入300ml甲醇攪拌30分鐘過濾,烘乾得白色固體的化合物1-5(5.8g,產率50.0%)。
1hnmr(400mhz,cdcl3,δ):
9.946-9.943(d,1h);8.996-8.976(d,1h);8.777-8.760(d,1h);8.423-8.388(t,4h);8.161(s,1h);7.910-7.811(m,5h);7.718-7.632(m,6h);7.595-7.558(t,1h);7.526-7.476(m,5h);7.439-7.382(m,2h)。
合成例3(化合物1-6的合成)
將4-二苯基-4-基-6-(4-溴苯基)-2-吡啶-3-基-嘧啶(4-biphenyl-4-yl-6-(4-bromophenyl)-2-pyridin-3-yl-pyrimidine)(10g,215mmol)與二苯並呋喃-4-硼酸(dibenzofuran-4-boronicacid)(5.48g,258mmol)放入反應瓶中,再加入甲苯100毫升,再將k2co3(4.46g,323mmol)溶解於40毫升的去離子水中加入反應瓶內,加入pd(pph3)4(0.371g,0.3mmol)再加入酒精20毫升,攪拌及於80℃回流反應16小時。反應完後加入150毫升去離子水攪拌至室溫,過濾出固體並以去離子水跟丙酮清洗,該固體再加入200毫升去離子水、50毫升甲醇及50毫升丙酮的混合溶液中攪拌30分鐘,再過濾,重複2次。烘乾該固體,再以1000ml甲苯加熱溶解後,通過矽膠柱,蒸餾濃縮後加入300ml甲醇攪拌30分鐘過濾,烘乾得白色固體的化合物1-6(7.2g,產率60.6%)。
1hnmr(400mhz,cdcl3,δ):
9.965-9.961(s,1h);9.028-8.998(d,1h);8.784-8.768(d,1h);8.507-8.486(d,2h);8.424-8.403(d,2h);8.208(s,1h);8.177-8.155(d,2h);8.037-7.997(t,2h);7.842-7.821(d,2h);7.733-7.705(d,3h);7.658-7.637(d,1h);7.530-7.475(m,5h);7.439-7.377(m,2h)。
合成例4(化合物1-7的合成)
將4-二苯基-4-基-6-(4-溴苯基)-2-吡啶-3-基-嘧啶(4-biphenyl-4-yl-6-(4-bromophenyl)-2-pyridin-3-yl-pyrimidine)(10g,215mmol)與二苯並噻吩-4-硼酸(dibenzothiophene-4-boronicacid)(5.89g,258mmol)放入反應瓶中,再加入甲苯100毫升,再將k2co3(4.46g,323mmol)溶解於40毫升的去離子水中加入反應瓶內,加入pd(pph3)4(0.371g,0.3mmol)再加入酒精20毫升,攪拌及於80℃回流反應16小時。反應完後加入150毫升去離子水攪拌至室溫,過濾出固體並以去離子水跟丙酮清洗,該固體再加入200毫升去離子水、50毫升甲醇及50毫升丙酮的混合溶液中攪拌30分鐘,再過濾,重複2次。烘乾該固體,再以1000ml甲苯加熱溶解後,通過矽膠柱,蒸餾濃縮後加入300ml甲醇攪拌30分鐘過濾,烘乾得白色固體的化合物1-7(7.1g,產率58.0%)。
1hnmr(400mhz,cdcl3,δ):
9.965-9.959(d,1h);9.015-8.996(m,1h);8.787-8.772(m,1h);8.469-8.407(m,4h);8.231-8.203(m,3h);7.995-7.974(d,2h);7.883-7.822(m,3h);7.726-7.703(d,2h);7.641-7.578(m,2h);7.531-7.403(m,6h)。
合成例5(化合物1-10的合成)
將4-二苯基-4-基-6-(4-溴苯基)-2-吡啶-3-基-嘧啶(4-biphenyl-4-yl-6-(4-bromophenyl)-2-pyridin-3-yl-pyrimidine)(10g,215mmol)與4-(1-苯基-1h-苯並[d]咪唑-2-基)苯基硼酸(4-(1-phenyl-1h-benzo[d]imidazol-2-yl)phenylboronicacid(8.12g,258mmol))放入反應瓶中,再加入甲苯100毫升,再將k2co3(4.46g,323mmol)溶解於40毫升的去離子水中加入反應瓶內,加入pd(pph3)4(0.371g,0.3mmol)再加入酒精20毫升,攪拌及於80℃回流反應16小時。反應完後加入150毫升去離子水攪拌至室溫,過濾出固體並以去離子水跟丙酮清洗,該固體再加入200毫升去離子水、50毫升甲醇及50毫升丙酮的混合溶液中攪拌30分鐘,再過濾,重複2次。烘乾該固體,再以1000ml甲苯加熱溶解後,通過矽膠柱,蒸餾濃縮後加入300ml甲醇攪拌30分鐘過濾,烘乾得白色固體的化合物1-10(3.1g,產率43.0%)。
1hnmr(400mhz,cdcl3,δ):
9.931(s,1h);8.992-8.969(d,1h);8.757(d,1h);8.369-8.362(d,4h);8.148(s,1h);7.929-7.910(d,1h);7.822-7.802(d,4h);7.729-7.644(m,7h);7.565-7.487(m,7h);7.433-7.386(m,4h)。
合成例6(化合物2-6的合成)
將4-二苯基-4-基-6-(4-溴苯基)-2-吡啶-4-基-嘧啶(4-biphenyl-4-yl-6-(4-bromophenyl)-2-pyridin-4-yl-pyrimidine)(10g,215mmol)與二苯並呋喃-4-硼酸(dibenzofuran-4-boronicacid)(5.48g,258mmol)放入反應瓶中,再加入甲苯100毫升,再將k2co3(4.46g,323mmol)溶解於40毫升的去離子水中加入反應瓶內,加入pd(pph3)4(0.371g,0.3mmol)再加入酒精20毫升,攪拌及於80℃回流反應16小時。反應完後加入150毫升去離子水攪拌至室溫,過濾出固體並以去離子水跟丙酮清洗,該固體再加入200毫升去離子水、50毫升甲醇及50毫升丙酮的混合溶液中攪拌30分鐘,再過濾,重複2次。烘乾該固體,再以1000ml甲苯加熱溶解後,通過矽膠柱,蒸餾濃縮後加入300ml甲醇攪拌30分鐘過濾,烘乾得白色固體的化合物2-6(9.6g,產率80.9%)。
1hnmr(400mhz,cdcl3,δ):
8.868-8.853(d,2h);8.617-8.602(d,2h);8.514-8.487(d,2h);8.424-8.403(d,2h);8.248(s,1h);8.187-8.160(d,2h);8.038-8.005(t,2h);7.852-7.825(d,2h);7.734-7.703(m,3h);7.656-7.637(d,1h);7.534-7.478(m,4h);7.448-7.380(m,2h)。
合成例7(化合物2-10的合成)
將4-二苯基-4-基-6-(4-溴苯基)-2-吡啶-4-基-嘧啶(4-biphenyl-4-yl-6-(4-bromophenyl)-2-pyridin-4-yl-pyrimidine)(10g,215mmol)與4-(1-苯基-1h-苯並[d]咪唑-2-基)苯基硼酸(4-(1-phenyl-1h-benzo[d]imidazol-2-yl)phenylboronicacid)(8.12g,258mmol)放入反應瓶中,再加入甲苯100毫升,再將k2co3(4.46g,323mmol)溶解於40毫升的去離子水中加入反應瓶內,加入pd(pph3)4(0.371g,0.3mmol)再加入酒精20毫升,攪拌及於80℃回流反應16小時。反應完後加入150毫升去離子水攪拌至室溫,過濾出固體並以去離子水跟丙酮清洗,該固體再加入200毫升去離子水、50毫升甲醇及50毫升丙酮的混合溶液中攪拌30分鐘,再過濾,重複2次。烘乾該固體,再以1000ml甲苯加熱溶解後,通過矽膠柱,蒸餾濃縮後加入300ml甲醇攪拌30分鐘過濾,烘乾得白色固體的化合物2-10(11g,產率78.0%)。
1hnmr(400mhz,cdcl3,δ):
δ8.847-8.833(d,2h);8.574-8.559(d,2h);8.390-8.359(m,4h);8.168(s,1h);7.932-7.912(d,1h);7.823-7.791(m,4h);7.730-7.687(t,4h);7.656-7.635(d,2h);7.586-7.486(m,5h);7.436-7.346(m,4h);7.314-7.258(t,2h)。
合成例8(化合物12-4的合成)
將4-二苯基-4-基-6-(3-溴苯基)-2-吡啶-3-基-嘧啶(4-biphenyl-4-yl-6-(3-bromophenyl)-2-pyridin-3-yl-pyrimidine)(10g,215mmol)與(3-(3-吡啶基)苯基硼酸(3-(3-pyridyl)phenylboronicacid)(5.14g,258mmol)放入反應瓶中,再加入甲苯100毫升,再將k2co3(4.46g,323mmol)溶解於40毫升的去離子水中加入反應瓶內,加入pd(pph3)4(0.371g,0.3mmol)再加入酒精20毫升,攪拌及於80℃回流反應16小時。反應完後加入150毫升去離子水攪拌至室溫,過濾出固體並以去離子水跟丙酮清洗,該固體再加入200毫升去離子水、50毫升甲醇及50毫升丙酮的混合溶液中攪拌30分鐘,再過濾,重複2次。烘乾該固體,再以1000ml甲苯加熱溶解後,通過矽膠柱,蒸餾濃縮後加入300ml甲醇攪拌30分鐘過濾,烘乾得白色固體的化合物12-4(8.7g,產率75.0%)。
1hnmr(400mhz,cdcl3,δ):
9.933-9.937(s,1h);8.987-8.950(t,2h);8.766-8.755(d,1h);8.644-8.632(d,1h);8.531(s,1h);8.403-8.382(d,2h);8.314-8.294(d,1h);8.167(s,1h);7.987-7.966(d,1h);7.888(s,1h);7.847-7.638(m,9h);7.518-7.400(m,5h)。
合成例9(化合物12-6的合成)
將4-二苯基-4-基-6-(3-溴苯基)-2-吡啶-3-基-嘧啶(4-biphenyl-4-yl-6-(3-bromophenyl)-2-pyridin-3-yl-pyrimidine)(10g,215mmol)與二苯並呋喃-4-硼酸(dibenzofuran-4-boronicacid)(5.48g,258mmol)放入反應瓶中,再加入甲苯100毫升,再將k2co3(4.46g,323mmol)溶解於40毫升的去離子水中加入反應瓶內,加入pd(pph3)4(0.371g,0.3mmol)再加入酒精20毫升,攪拌及於80℃回流反應16小時。反應完後加入150毫升去離子水攪拌至室溫,過濾出固體並以去離子水跟丙酮清洗,該固體再加入200毫升去離子水、50毫升甲醇及50毫升丙酮的混合溶液中攪拌30分鐘,再過濾,重複2次。烘乾該固體,再以1000ml甲苯加熱溶解後,通過矽膠柱,蒸餾濃縮後加入300ml甲醇攪拌30分鐘過濾,烘乾得白色固體的化合物12-6(8.1g,產率68.0%)。
1hnmr(400mhz,cdcl3,δ):
9.970(d,1h);9.022-8.992(d,1h);8.848-8.840(t,1h);8.770-8.761(d,1h);8.415-8.394(d,2h);8.362-8.342(d,1h);8.209(s,1h);8.139-8.121(d,1h);8.046-8.008(t,2h);7.824-7.641(m,4h);7.534-7.466(m,5h);7.431-7.378(m,2h)。
合成例10(化合物12-10的合成)
將4-二苯基-4-基-6-(3-溴苯基)-2-吡啶-3-基-嘧啶(4-biphenyl-4-yl-6-(3-bromophenyl)-2-pyridin-3-yl-pyrimidine)(10g,215mmol)與4-(1-苯基-1h-苯並[d]咪唑-2-基)苯基硼酸(4-(1-phenyl-1h-benzo[d]imidazol-2-yl)phenylboronicacid)(8.12g,258mmol)放入反應瓶中,再加入甲苯100毫升,再將k2co3(4.46g,323mmol)溶解於40毫升的去離子水中加入反應瓶內,加入pd(pph3)4(0.371g,0.3mmol)再加入酒精20毫升,攪拌及於80℃回流反應16小時。反應完後加入150毫升去離子水攪拌至室溫,過濾出固體並以去離子水跟丙酮清洗,該固體再加入200毫升去離子水、50毫升甲醇及50毫升丙酮的混合溶液中攪拌30分鐘,再過濾,重複2次。烘乾該固體,再以1000ml甲苯加熱溶解後,通過矽膠柱,蒸餾濃縮後加入300ml甲醇攪拌30分鐘過濾,烘乾得白色固體的化合物12-10(7.8g,產率55.4%)。
1hnmr(400mhz,cdcl3,δ):
9.925(s,1h);8.977-8.959(d,1h);8.767-8.757(d,1h);8.490(s,1h);8.398-8.377(d,1h);8.272-8.251(d,1h);8.140(s,1h);7.931-7.911(d,1h);7.823-7.776(t,3h);7.748-7.631(m,8h);7.583-7.471(m,7h);7.433-7.346(m,4h)。
合成例11(化合物23-6的合成)
將4-二苯基-4-基-6-(3,5-二溴苯基)-2-吡啶-3-基-嘧啶(4-biphenyl-4-yl-6-(3,5-dibromo-phenyl)-2-pyridin-3-yl-pyrimidine(10g,184mmol))與二苯並呋喃-4-硼酸(dibenzofuran-4-boronicacid)(8.6g,406mmol)放入反應瓶中,再加入甲苯100毫升,再將k2co3(4.46g,323mmol)溶解於40毫升的去離子水中加入反應瓶內,加入pd(pph3)4(0.371g,0.3mmol)再加入酒精20毫升,攪拌及於80℃回流反應16小時。反應完後加入150毫升去離子水攪拌至室溫,過濾出固體並以去離子水跟丙酮清洗,該固體再加入200毫升去離子水、50毫升甲醇及50毫升丙酮的混合溶液中攪拌30分鐘,再過濾,重複2次。烘乾該固體,再以1000ml甲苯加熱溶解後,通過矽膠柱,蒸餾濃縮後加入300ml甲醇攪拌30分鐘過濾,烘乾得白色固體的化合物23-6(7.2g,產率54.4%)。
1hnmr(400mhz,cdcl3,δ):
δ10.009-10.003(d,1h);9.053-9.033(d,1h);8.887-8.883(s,2h);8.778-8.766(d,1h);8.627(t,1h);8.440-8.418(d,2h);8.302(s,1h);8.064-8.045(d,1h);7.854-7.807(m,4h);7.707-7.651(m,4h);7.572-7.469(m,7h);7.423-7.387(m,3h)。
合成例12(化合物23-8的合成)
將4-二苯基-4-基-6-(3,5-二溴苯基)-2-吡啶-3-基-嘧啶(4-biphenyl-4-yl-6-(3,5-dibromo-phenyl)-2-pyridin-3-yl-pyrimidine(10g,184mmol))與萘-1-基硼酸(naphthalen-1-ylboronicacid(7.9g,459mmol))放入反應瓶中,再加入甲苯100毫升,再將k2co3(4.46g,323mmol)溶解於40毫升的去離子水中加入反應瓶內,加入pd(pph3)4(0.371g,0.3mmol)再加入酒精20毫升,攪拌及於80℃回流反應16小時。反應完後加入150毫升去離子水攪拌至室溫,過濾出固體並以去離子水跟丙酮清洗,該固體再加入200毫升去離子水、50毫升甲醇及50毫升丙酮的混合溶液中攪拌30分鐘,再過濾,重複2次。烘乾該固體,再以1000ml甲苯加熱溶解後,通過矽膠柱,蒸餾濃縮後加入300ml甲醇攪拌30分鐘過濾,烘乾得白色固體的化合物23-8(11g,產率94.0%)。
1hnmr(400mhz,cdcl3,δ):
9.905(d,1h);8.928-8.923(d,1h);8.720(d,1h);8.503(s,2h);8.383-8.352(d,2h);8.192(s,1h);8.123-8.103(d,2h);7.977-7.941(t,4h);7.869(d,1h);7.790-7.759(d,2h);7.684-7.380(m,14h)。
合成例13(化合物36-1的合成)
將4-二苯基-4-基-6-(3,5-二溴苯基)-2-吡啶-3-基-嘧啶(4-biphenyl-4-yl-6-(3,5-dibromo-phenyl)-2-pyridin-3-yl-pyrimidine(10g,184mmol))、咔唑carbazole(7.1g,424mmol)與stb(13.116g,136mmol)放入反應瓶中,再加入甲苯150毫升並攪拌,再加入pd(dba)2(0.673g,1.17mmol)與p(t-butyl)3(0.662g,0.312mmol)並攪拌及於115℃回流反應18小時。反應完後加入150毫升去離子水攪拌至室溫,過濾出固體並以去離子水跟丙酮清洗,該固體再加入200毫升去離子水、50毫升甲醇及50毫升丙酮的混合溶液中攪拌30分鐘,再過濾,重複2次。烘乾該固體,再以1000ml甲苯加熱溶解後,通過矽膠柱,蒸餾濃縮後加入300ml甲醇攪拌30分鐘過濾,烘乾得黃色固體的化合物36-1(6.3g,產率48.0%)。
1hnmr(400mhz,cdcl3,δ):
9.898-9.894(d,1h);8.940-8.910(d,1h);8.752-8.736(d,1h);8.640-8.635(d,2h);8.380-8.354(d,2h);8.219-8.200(d,4h);8.163(s,1h);8.038-8.028(t,1h);7.799-7.768(d,2h);7.682-7.631(m,6h);7.519-7.345(m,12h)。
實施例1(有機電致發光元件的製造)
於基底載入蒸鍍系統使用前,先以溶劑及紫外線臭氧清洗基底進行脫脂。之後,將基底傳送至真空沉積室,於基底的頂部沉積所有層。由加熱的蒸鍍舟(boat)在約10-6託的真空度依序沉積圖2所示的各層:
a)空穴注入層,厚度20納米,hat-cn;
b)空穴傳輸層,厚度60納米,ht;
c)發光層,厚度30納米,包含摻雜有3%體積比bd的bh,(bh和bd為中國臺灣昱鐳光電科技股份有限公司的商品名);
d)電子傳輸層,厚度25納米,包含化合物1-4及摻雜的喹啉鋰(liq);
e)電子注入層,厚度1納米,氟化鋰(lif);及
f)陰極,厚度約150納米,包含a1。
元件結構可表示如:ito/hat-cn(20納米)/ht(60納米)/bh-3%bd(30納米)/化合物1-4:liq(25納米)/氟化鋰(1納米)/a1(150納米)。
於沉積形成上述各層後,該元件自沉積室傳送至乾燥箱中,隨即以uv可固化環氧樹脂及含有吸溼劑的玻璃蓋板進行封裝。該有機電致發光元件具有3平方毫米的發光區域。於連接外部電源後,該有機電致發光元件於直流電壓下運作,其所發光性質確認於後列表1。
所有製成的有機電致發光元件的電致發光性質均使用定電流源(keithley2400sourcemeter,madebykeithleyinstruments,inc.,cleveland,ohio)及光度計(photoresearchspectrascanpr650,madebyphotoresearch,inc.,chatsworth,calif.)於室溫下進行測量。
藉由驅動定電流依據發光層的光色於室溫及不同初始發光度來測試元件的使用壽命(或稱穩定性)。光色使用國際照明委元會所定cie坐標表示。
除將實施例1中電子傳輸層的化合物1-4各別置換為化合物1-6、1-7、12-6、23-8及36-1,實施例2、實施例3、實施例4、實施例5及實施例6如實施例1的層結構。
比較例1(有機電致發光元件的製造)
除將實施例1中電子傳輸層的化合物1-4置換為et(中國臺灣昱鐳光電科技股份有限公司),比較例1的製造近似於實施例1的層結構。比較例1的元件結構可表示如:ito/hat-cn(20納米)/ht(60納米)/bh-3%bd(30納米)/et:liq(20納米)/lif(1納米)/a1(150納米)。
製成的有機電致發光元件的發光的波峰波長、最大發光效率、驅動電壓及穩定性列示於表1。圖4為製成的有機電致發光元件的電致發光光譜。
表1
本發明不限於上述實施例、方法及實例,以請求保護本發明的範圍及精神內所有實施例及方法為準。
實用性
如上所述,包含適用於有機電致發光元件材料的本發明的有機電致發光元件可實現長使用壽命的特性,並具有高發光效率和維持低的驅動電壓。因此,本發明有機電致發光元件具有極高的技術價值且適用於平面顯示器、行動通信元件的顯示器、利用其為面發光體特性的光源、記號板。